DL. (v. electrónica):
B-16965-2010
ISSN (v. electrónica):
2013-8415
4 números/año

2Unidad Oftalmología Pediátrica, Hospital Universitari Vall d’Hebron. Barcelona
3Servicio Oftalmología, Hospital Universitari Bellvitge. Barcelona
4Unidad Neurología Pediátrica, Hospital Universitari Vall d’Hebron. Barcelona
CORRESPONDENCIA
Servicio Oftalmología
Hospital Mútua de Terrassa. Barcelona
E-mail: saintgerons@gmail.com
RESUMEN
RESUM
ABSTRACT
Introducción
La atrofia óptica traduce la pérdida de axones de las células ganglionares de la retina secundaria a diferentes lesiones que afectan la vía óptica anterior. Menos frecuentemente un daño en la vía óptica retrogeniculada conduce a la atrofia por degeneración retrógrada transináptica. En la exploración se aprecia un disco óptico pálido de forma difusa o segmentaria, pero sin cambios en su tamaño. El aspecto del fondo del ojo no permite establecer su etiología en la mayoría de los casos.En niños mayores y colaboradores se puede realizar una exploración oftalmológica completa, lo que es muy valioso para establecer el diagnóstico (agudeza visual, visión de colores, fondo de ojo y campimetría). En los más pequeños y en los no colaboradores por su estado neurológico, el aspecto del fondo de ojo, la exploración sistémica y neurológica puede ser insuficiente, por lo que habrá que recurrir a estudios complementarios.
En este trabajo se revisan las principales causas de atrofia óptica en la edad pediátrica (Tabla 1), se discute el diagnóstico diferencial y se ha consensuado un protocolo diagnóstico con los participantes del Grup de Treball de Neuro-oftalmología de Barcelona.

Tabla 1. Etiología de la atrofia óptica en la infancia.

Etiología
Lesiones intracraneales compresivas / infiltrativas
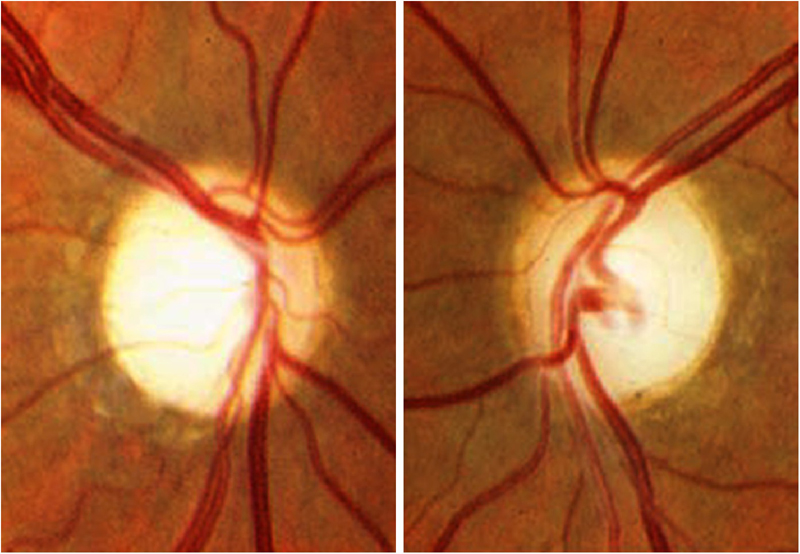
Los tumores supraselares congénitos pueden ser causa de atrofia óptica bilateral en los dos primeros años de vida. Estas lesiones intracraneales son craneofaringiomas, gliomas, tumores pituitarios no funcionantes, meningiomas, malformaciones arteriovenosas, aneurismas, tumores metastásicos y quistes aracnoideos. Rara vez se asocian a tumores como lipomas o lipodermoides. Cuando la compresión afecta al quiasma aparece la atrofia óptica en banda, con mayor palidez de los cuadrantes nasal y temporal y preservación de los cuadrantes superior e inferior (Figura 1). Estos niños, además de atrofia óptica pueden presentar nistagmo.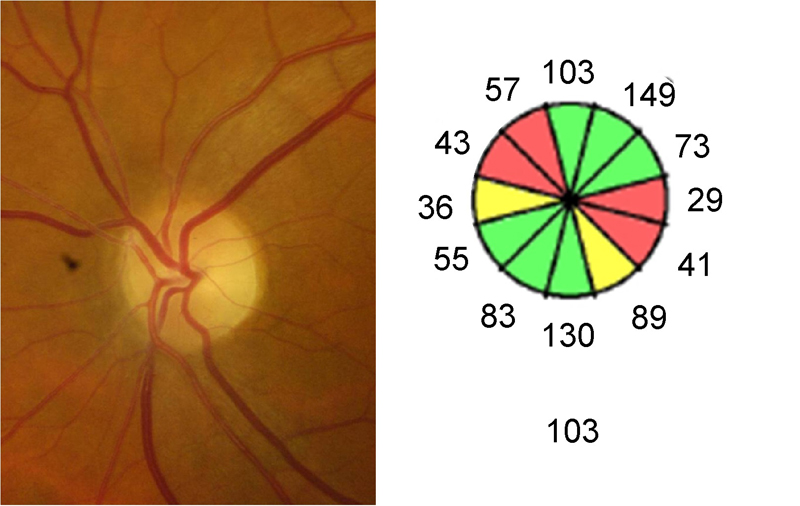
Figura 1. Palidez papilar en banda (atrofia óptica en pajarita). En la retinografía se aprecia mayor palidez en los sectores nasal y temporal del disco, mientras el cuadrante superior e inferior presenta una coloración normal del anillo neuro-retiniano. La OCT reproduce este patrón de pérdida de fibras en el la distribución por sectores horarios.
Craneofaringioma
El craneofaringioma es la tumoración benigna supraselar más frecuente en la edad pediátrica. Representa entre el 6 y el 10% de los tumores intracraneales de la infancia. Su origen es congénito y aparece a partir de restos de la bolsa de Rathke. La localización puede ser exclusivamente intraselar, supraselar o intrasupraselar; y son tumores de crecimiento lento. Es ligeramente más frecuente en el sexo masculino y se diagnostica con mayor frecuencia entre los 5 y 14 años de edad. Los pacientes experimentan pérdida visual progresiva por compresión de uno o ambos nervios ópticos, del quiasma o de las vías ópticas, y menos frecuente por papiledema crónico. En ocasiones la pérdida visual puede ser aguda.
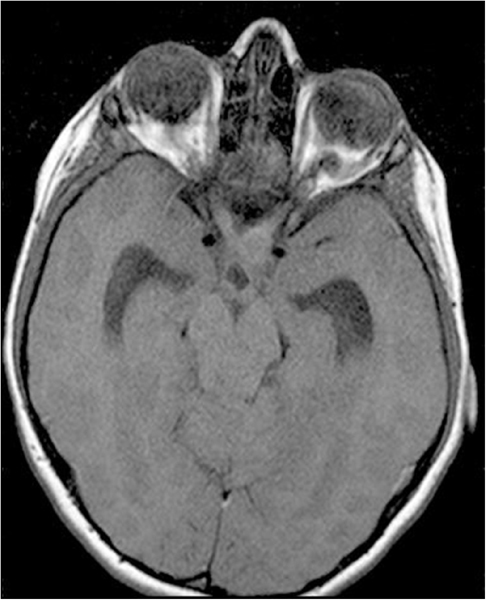
El craneofaringioma debe descartarse siempre en los casos de presunta pérdida visual psicógena y las ambliopías sin factores ambliogénicos como anisometropía o estrabismo, y atrofia óptica; por lo tanto está indicada una prueba de imagen. La tomografía computarizada (TC) muestra lesiones calcificadas quísticas en localización supraselar. Si el tumor es isodenso con el líquido cefaloraquídeo, puede únicamente aparecer distorsión de la cisterna supraselar. En la resonancia magnética (RM) los hallazgos dependerán de la composición del tumor: colesterol, metahemoglobina o malla trabecular ósea (Figura 2). Aunque histológicamente son tumores benignos, pueden tener un comportamiento agresivo por su localización y el diagnóstico diferencial se establece con el meningioma, el adenoma hipofisario, el disgerminoma, el quiste de la bolsa de Rathke y el quiste epidermoide supraselar.
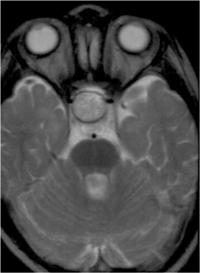
Figura 2. Craneofaringioma RMN T2. Lesión de grandes dimensiones y redondeada con contenido líquido por el componente quístico de localización supraselar.
Glioma del nervio óptico
La edad de presentación típica es alrededor de los 9 años y afecta a ambos sexos por igual. El 75% de los gliomas del nervio óptico se presentan en la primera década de la vida y el 90% en las dos primeras. La prevalencia de neurofibromatosis tipo 1 (NF-1) en pacientes con glioma de la vía óptica varía según las series entre el 10 y el 70%. En la NF-1 el glioma del nervio óptico puede ser multifocal y en ocasiones involucra ambos nervios (Figura 3).
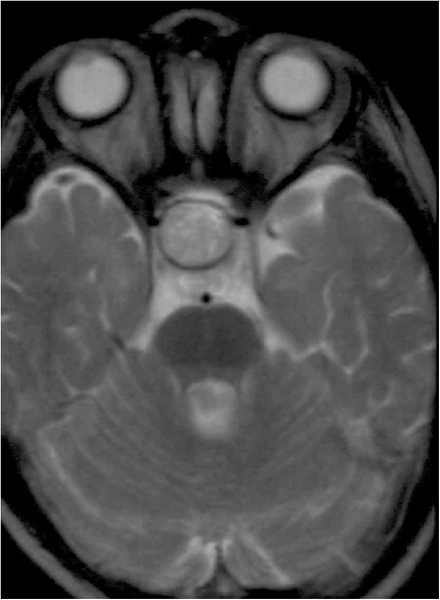
Figura 3. Glioma nervio óptico que invade quiasma RMN T1.
Adenoma hipofisario
Estos tumores son infrecuentes en la infancia y la mayor parte de ellos debutan después de la pubertad con frecuentes cefaleas y pérdida visual. El 70% son adenomas secretores en el momento de la presentación como fallo de maduración sexual, el prolactinoma es el más frecuente. Los adenomas hipofisarios tienen mayor extensión extraselar y mayor capacidad invasiva cuando aparecen en el período prepuberal.
Disgerminoma
Debe ser sospechado en un paciente con diabetes insípida con defectos del campo visual bitemporales.
Lesiones compresivas no tumorales infrecuentes que causan atrofia óptica
Osteopetrosis
Es una enfermedad ósea hereditaria metabólica que cursa con aumento de densidad ósea por la hipofunción de los osteoclastos. La atrofia óptica es secundaria al estrechamiento del canal óptico (compresión) o por hipertensión intracraneal (papiledema).
Craneosinostosis
Se caracteriza por el cierre precoz de una o más suturas craneales. El escaso desarrollo del canal óptico y la hipertensión intracraneal pueden provocar atrofia óptica por dos mecanismos diferentes, la compresión directa del nervio óptico y por papiledema evolucionado respectivamente.
Displasia fibrosa
Atrofia óptica post papiledema
Cuando el grado distensión de la cavidad craneal en niños con suturas abiertas es insuficiente para prevenir el aumento de la presión en la vaina del nervio óptico puede aparecer papiledema (Figura 4). Se han descrito casos de atrofia óptica post papiledema en niños menores de dos años, los mecanismos responsables del aumento de la presión intracraneal y secundariamente un papiledema son:- Aumento del tejido intracraneal por lesión ocupante de espacio.
- Aumento del volumen tisular por edema cerebral difuso o focal.
- Aumento de la producción de líquido cefalo-raquídeo por un tumor intracraneal (ependimoma).
- Reducción del volumen total en la bóveda craneal por engrosamiento craneal.
- Bloqueo de la circulación del líquido cefaloraquídeo en el sistema ventricular (hidrocefalia obstructiva o no comunicante) o en las granulaciones aracnoideas (hidrocefalia comunicante o no obstructiva).
- Reducción en la absorción del líquido cefaloraquídeo por obstrucción (trombosis senos venosos durales) o compromiso del flujo venoso.
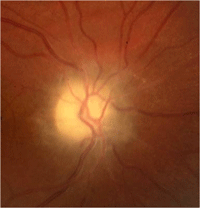
Figura 4. Atrofia óptica post papiledema. La retinografía revela palidez papilar con márgenes del disco poco definidos, así como hialinización de los vasos que emergen de disco.
Causas no compresivas de atrofia óptica en contexto de patología tumoral
Este apartado engloba una miscelánea, los efectos de los tratamientos antitumorales (radioterapia y quimioterápicos) y los síndromes paraneoplásicos.Los pacientes que reciben una dosis de radiación mayor a 50-60 Gy o dosis superiores a 200 cGy al día tienen un elevado riesgo de desarrollar efectos indeseables. La neuropatía por irradiación puede presentarse de forma precoz inmediata por edema, precoz diferida por desmielinización al cabo de unas semanas, o de forma tardía tras meses o años por isquemia y necrosis.
Es infrecuente la degeneración paraneoplásica axonal en la infancia, la retinopatía paraneoplásica es más frecuente en adultos. En cuanto en a la toxicidad por antineoplásicos se discute más adelante en el apartado de neuropatías ópticas tóxicas.
Hipoxia-isquemia
La valoración del daño hipóxico-isquémico perinatal en el sistema visual es no es una tarea sencilla, ya que puede existir tanto palidez como hipoplasia del nervio óptico o ambas. Si el daño sobre el sistema visual se presenta en fases precoces de la gestación, cuando el nervio no está completamente desarrollado, el disco óptico aparece pequeño y con palidez variable (hipoplasia). La deficiencia visual cerebral se puede acompañar de cierto grado de palidez papilar por degeneración transináptica. Los niños con hemorragia intraventricular grave (grado III y IV) con frecuencia presentan atrofia óptica con discos de dimensiones normales porque la muerte de los axones sucede tras el desarrollo completo del nervio óptico.Encefalopatía hipóxico-isquémica
Es el daño difuso o focal cerebral secundario a un estado de asfixia perinatal y a los sucesivos trastornos fisiopatológicos. En el sistema nervioso cerebral la distribución del daño depende del aporte vascular preferencial en situaciones de hipoxia grave, y éste de la madurez cerebral, por tanto, de la edad gestacional. El patrón de flujo arterial se modifica a partir de las 36 semanas de gestación; antes de esta fecha los vasos tienen una dirección preferencial desde la corteza hacia la sustancia blanca periventricular (flujo ventriculopetal), y tras la semana 36, el flujo preferencial adquiere una dirección más periférica (hacia la sustancia blanca subcortical y corteza parasagital). En consecuencia, si la asfixia se produce antes de la 35 semana de edad gestacional, el daño cerebral se localiza en la sustancia blanca periventricular, mientras que si se produce de forma más tardía, afecta la corteza parietal parasagital.Neuropatia óptica isquémica
La neuropatía óptica isquémica anterior no arterítica es muy poco frecuente en la infancia. Se clasifica en anterior o posterior en función de la localización de la isquemia del nervio óptico. Si aparece edema de papila se etiqueta de neuropatía óptica isquémica anterior, y si el fondo es normal en el momento de la presentación, de neuropatía óptica isquémica posterior. En ambos casos, la evolución es a la atrofia óptica, uni o bilateral. Es una complicación de un tratamiento antihipertensivo agresivo, de cirugía espinal, diálisis peritoneal y con el uso de sildenafilo en la hipertensión pulmonar. También aparece en pacientes con migraña, y con alteraciones protrombóticas. La neuropatía óptica isquémica posterior se ha descrito en niños con shock cardiovascular e hipotensión debida a la pérdida aguda de sangre.Neuropatía óptica traumática
El mecanismo de producción puede ser directo o indirecto. En el primer caso el traumatismo provoca una disrupción de la anatomía del nervio óptico, y se asocia a heridas penetrantes del ojo o de la órbita. En traumatismos cerrados, el mecanismo es indirecto y se atribuye a la transmisión de las fuerzas dinámicas hacia el nervio óptico desde una localización a distancia sin disrupción de la anatomía del mismo. La porción intracanalicular es la más vulnerable en estos casos pues la dura está íntimamente adherida al periostio. Inicialmente, el aspecto del nervio óptico es normal en los traumatismos indirectos que afecten la porción intracanalicular o intracraneal y hacia las seis semanas aparecerá la atrofia óptica.Atrofias ópticas hereditarias
Es un grupo heterogéneo de enfermedades que se manifiesta con atrofia óptica bilateral y transmisión genética. La forma de presentación más frecuente es pérdida visual central, bilateral y simétrica, y discromatopsia. Un hallazgo común es la afectación del haz papilo-macular provocando defectos centrales o centrocecales del campo visual. El déficit visual es permanente y progresivo en muchas enfermedades. En el diagnóstico diferencial deben excluirse enfermedades retinianas primarias que causan atrofia óptica secundariamente. Es muy importante distinguir las enfermedades en las cuales la neuropatía óptica es la afectación principal (con o sin hallazgos sistémicos o neurológicos), de las enfermedades sistémicas o neurológicas en las cuales puede haber afectación del nervio óptico.Atrofia óptica dominante
Es la forma más frecuente de atrofia óptica hereditaria. La pérdida visual tiene un inicio insidioso en la primera década de la vida y se pone de manifiesto de manera solapada o casual. La agudeza visual varía de 20/70 a 20/100, aunque puede ser de 20/20 o de cuenta dedos. También presenta variabilidad intraindividual (visiones asimétricas) y entre los miembros de la misma familia. Los hallazgos campimétricos son: defectos centrales, centrocecales, paracentrales o hemianopsia superotemporal bilateral.El fondo de ojo muestra discos ópticos con palidez temporal o atrofia completa, en algunos casos hay una excavación focal temporal. La pérdida del haz papilomacular es el hallazgo más precoz, algunos pacientes presentan cambios sutiles en el pigmento macular (Figura 5a y Figura 5b).
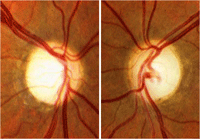
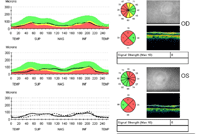
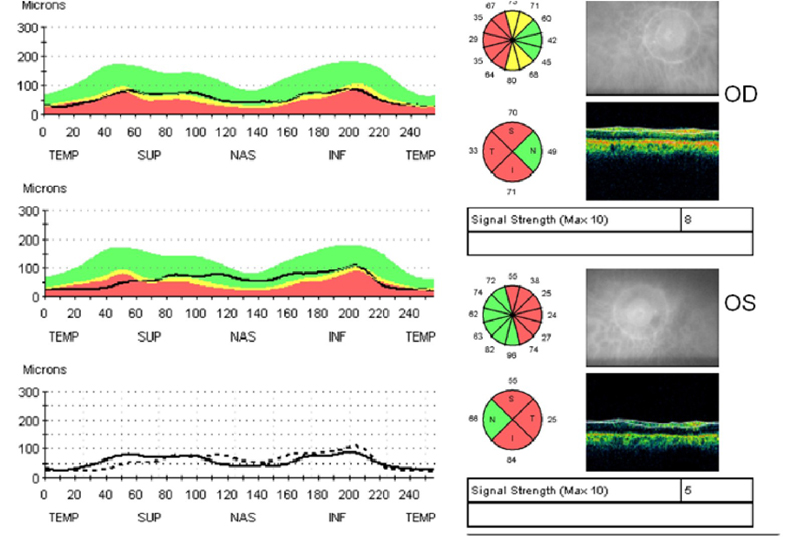
Figura 5. Atrofia óptica dominante OPA positiva. Se aprecia palidez del disco óptico y del anillo neuro-retiniano con excavación. (5a) OCT RNFL Stratus. Hay pérdida de la capa de fibras nerviosas en la porción temporal con preservación de los cuadrantes nasales (5b).
La mayoría de pacientes con atrofia óptica dominante son monosintomáticos pero hay raras excepciones en las cuales se asocia a retraso mental, hipoacusia y oftalmoplegía externa progresiva. (OPA-plus) La atrofia óptica dominante es genéticamente heterogénea, el gen OPA1 es el gen más frecuentemente origen de las mutaciones. También, se han identificado otros genes implicados: OPA3, OPA4 y OPA5.
Neuropatía óptica de leber
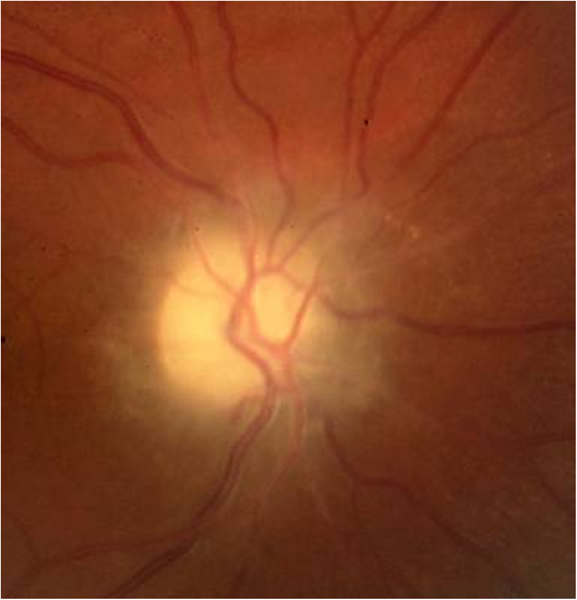
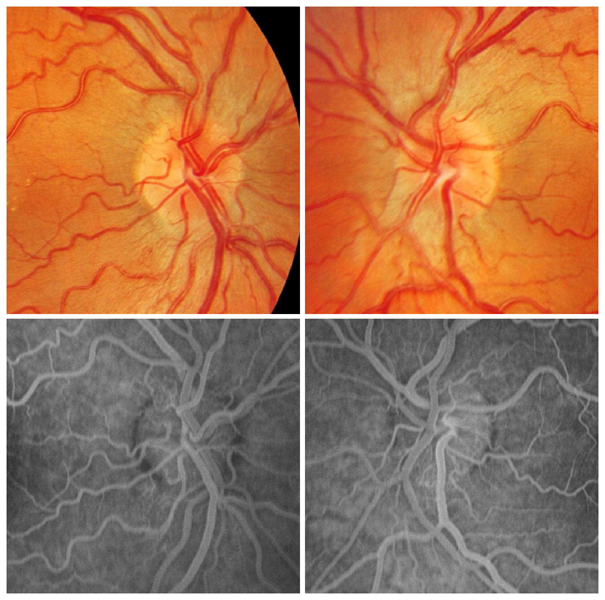
Está producida por diversas mutaciones del DNA mitocondrial y es de herencia materna. El 95% de los casos, las mutaciones se hallan en los siguientes puntos G11778A (50-76%), G3460A (7-30%) y T14484C (7-30%).Se manifiesta más frecuentemente entre los 15-35 años, el rango de edad descrito oscila entre 1 y 80 años. Aparece con visión borrosa unilateral que progresa rápidamente y afecta al ojo contralateral en días o meses. La agudeza visual se estabiliza en 20/200 o inferior con un rango de 20/40 a no percepción luminosa. El defecto de campo visual típico es central o centro-cecal, en algunos pacientes puede extenderse hacia el campo superior. La tríada característica del fondo de ojo previa al episodio agudo es la siguiente: microangiopatía telangiectásica peripapilar, pseudoedema de la papila y de la capa de fibras peripapilar y ausencia de fuga en la angiografía fluoresceínica, aunque no todos los pacientes presentan estos cambios (Figura 6).
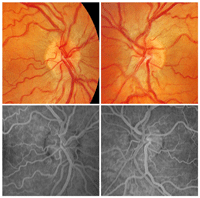
Figura 6. Neuropatía óptica hereditaria de Leber. En fase aguda aparecen telangiectasias peripapilares y tumefacción del disco óptico secundario por el aumento de tamaño de los axones de las células ganglionares de la retina Como no hay fuga de contraste en la angiografía fluoresceínica, este fenómeno se conoce como pseudoedema y se atribuye a la microangiopatía de la retina.
Atrofia óptica recesiva
Este es un grupo heterogéneo y por una parte se incluyen enfermedades como la atrofia óptica de Behr y el síndrome de Costeff, así como otras anomalías neurológicas en las que la atrofia óptica es una de sus manifestaciones. Por otra parte existe la forma aislada, monosintomática y recesiva de atrofia óptica. Es muy poco frecuente y se presenta en el nacimiento, o bien en los primeros años de vida. La visión es muy pobre (inferior a 20/200) existe acromatopsia o discromatopsia severa y se asocia a nistagmo por la baja visión. El campo visual presenta constricción con escotomas paracentrales y el disco óptico marcada atrofia difusa con una excavación profunda. Antes de establecer el diagnóstico de atrofia óptica recesiva deben excluirse otras enfermedades más frecuentes que causen atrofia óptica, por lo que es necesaria la exploración completa de la retina y un estudio electro-retinográfico (ERG) normal.Atrofia óptica ligada al cromosoma X
Se caracteriza por afectación severa de la agudeza visual en los primeros años de vida que progresa lentamente, alteración de la visión cromática y atrofia óptica sin nistagmo. Puede haber alteraciones neurológicas sutiles como ausencia de respuesta plantar, temblor e inestabilidad vesical.Síndrome de Behr
Es una variante de la atrofia óptica recesiva que también debuta en edades tempranas y se asocia a otras anomalías como la ataxia, disfunción piramidal y extrapiramidal, hipertonía, paresia juvenil espástica, retraso mental, incontinencia urinaria y pie cavo. El diagnóstico debe considerarse en pacientes con ataxia familiar y atrofia óptica.Síndrome de Wolfram (DIDMOAD)
El síndrome de Wolfram es una enfermedad neurodegenerativa de herencia autosómica recesiva. Se produce por la mutación en el gen WFS1. Se caracteriza por diabetes central insípida, diabetes mellitus, atrofia óptica y sordera. Otras anomalías incluyen: ptosis, braquidactilia, anosmia, ataxia, nistagmo, epilepsia, retraso mental, alteraciones psiquiátricas, aumento de las proteínas en el líquido cefaloraquídeo, talla baja, enfermedades cardíacas congénitas, miocarditis y anomalías genitourinarias. Las diferentes manifestaciones del síndrome están temporalmente separadas, la diabetes mellitus suele ser la primera manifestación de esta enfermedad en la primera década de la vida, más tarde aparecen la neuropatía óptica y la hipoacusia. El diagnóstico de atrofia óptica se establece alrededor de los 12 años y la visión suele ser inferior a 20/200. También se han descrito casos de retinopatía pigmentaria y ERG anómalo en este síndrome.Atrofia óptica progresiva y sordera autosómica dominante
Estos pacientes presentan una sordera severa desde nacimiento, mientras que su disfunción visual es leve en edades tempranas y la visión prácticamente normal hasta los 24 años. Existe una gran variabilidad intrafamiliar en la edad de debut de los síntomas.Atrofia óptica sordera y oftalmoplegía
Se caracteriza por atrofia óptica, sordera, ptosis, oftalmoplegía, distaxia y miopatía. La pérdida visual se detecta alrededor de los 11 años. El ERG puede ser anómalo a pesar de la ausencia de cambios retinianos pigmentarios lo que sugiere una combinación de degeneración retiniana primaria y neuropatía óptica primaria.Atrofia óptica progresiva con sordera progresiva y ataxia autosómica dominante (Síndrome de CAPOS)
Se caracteriza por atrofia óptica, sordera, ataxia y debilidad de las extremidades. La edad de presentación de la pérdida visual varía entre los 2 y los 9 años. La sordera es moderada y lentamente progresiva. Se han descrito 3 familias con atrofia óptica, sordera, ataxia cerebelosa, arreflexia sin neuropatía periférica y pies cavos (Síndrome de CAPOS).Atrofia óptica hereditaria, sordera y polineuropatía
Estas alteraciones pueden ser incluidas dentro del espectro de polineuropatías hereditarias que incluye la enfermedad de Charcot-Marie-Tooth. El grado de sordera y atrofia óptica es variable.Atrofia óptica autosómica recesiva con sordera progresiva, cuadraplegía espástica, deterioro mental y muerte
El denominador común de esta enfermedad es una degeneración sistemática y selectiva de los sistemas ópticos, cocleares, dentados y del lemnisco medial. No hay afectación del los ganglios basales y el diagnóstico es clínico.Atrofia óptico-acústica con demencia
Existe una degeneración generalizada del sistema nervioso central con calcificación extensa.Enfermedades retinianas
Las enfermedades que afectan la capa de fibras ganglionares pueden conducir a la atrofia óptica, por lo que ésta representa un hallazgo tardío en muchas enfermedades retinianas degenerativas difusas. Las más representativas son las distrofias retinianas congénitas, degeneraciones tapetorretinianas, lipofuscinosis neuronal ceroidea (ej. enfermedad de Batten), retinopatías infecciosas e inflamatorias (ej. neurorretinitis difusa unilateral subaguda, retinitis por citomegalovirus, toxoplasmosis) y oclusión central o de rama arterial de la retina.La atenuación arteriolar en el fondo de ojo sugiere la presencia de una enfermedad retiniana. En aquellos casos de atrofia óptica con marcado adelgazamiento arteriolar está indicado practicar un estudio ERG.
Neuropatías ópticas tóxicas y nutricionales
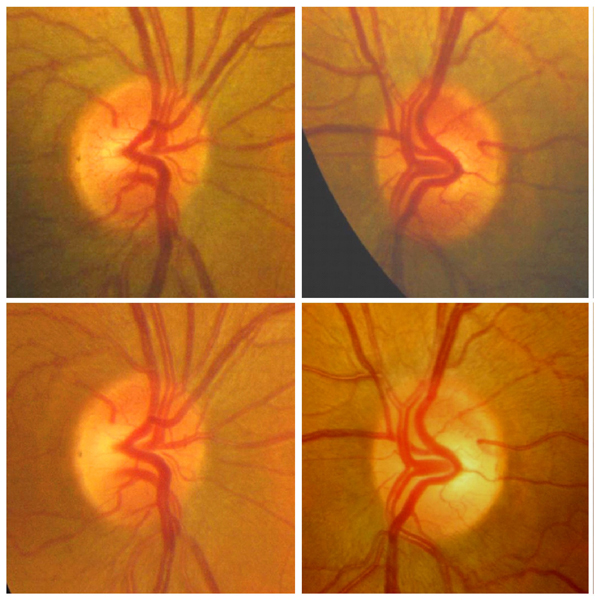
Este grupo de neuropatías producen una pérdida visual bilateral, simétrica, de inicio subagudo e indoloro. La discromatopsia es precoz y el defecto campimétrico suele ser central o centrocecal. Inicialmente, el fondo de ojo es normal, pero con el tiempo aparece palidez del disco óptico que predomina en el sector temporal del disco óptico (Figura 7). Para realizar un correcto diagnóstico, la historia clínica deber ser tomada de manera minuciosa, y centrada en los fármacos que toma el paciente y realizar una determinación de vitaminas (B1, B6, B12) y ácido fólico en sangre.
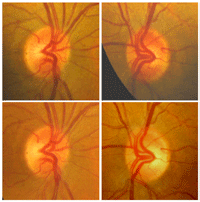
Figura 7. Neuropatía óptica tóxico nutricional. En la fase inicial el disco óptico aparece normal o existe una discreta hiperemia (arriba) En fase evolucionada, desaparecen estos fenómenos y se observa cierta palidez del cuadrante temporal en grado variable (abajo).
Causas:
-
Nutricionales
- Déficit tiamina, cobalamina, piridoxina, ácido fólico y riboflavina.
- Se debe sospechar en casos de malnutrición por dietas restrictivas o adolescentes con anorexia y condiciones con malabsorción gastrointestinal.
-
Tóxicas
- Etambutol: dosis ≥25 mg/Kg/día presentan un riesgo de neuropatía óptica del 5-6%. En casos de insuficiencia renal puede aparecer toxicidad a dosis menores porque este fármaco tiene una eliminación renal
- Isoniacida.
- Cloranfenicol: actúa como un inhibidor específico de la síntesis proteica mitocondrial en pacientes con fibrosis quística.
- Rifampicina.
- Linezolid.
- Penicilamina.
- Melatonina.
- Amiodarona.
- Quimioterápicos: vincristina, clorambucil, cisplatino, citosina arabinósido, 5-fluorouracilo, interferon, metotrexato y carmustina.
- Inmunosupresores: ciclosporina A y tacrolimús.
- Metanol.
- Plomo.
- Cobalto.
La vigabatrina causa retinopatía que secundariamente se traduce en una afectación del nervio óptico. El 30-40% de los pacientes presentan una reducción del campo visual periférico, que suele ser asintomático. En los estudios realizados con tomografía de coherencia óptica de capa de fibras nerviosas peripapilares, se ha objetivado un adelgazamiento en el sector nasal y preservación del sector temporales.
---------------------------------
*La segunda parte de este artículo se publicará en Annals d’Oftalmologia 2014;22(1).