Volumen 22 - Número 5 - Diciembre 2014
Tomografía de coherencia óptica (OCT) en las distrofias retinianas
MS. Alforja Castiella, J. Giralt Josa, R. Casaroli Marano
Unidad de Vítreo-Retina. Institut Clínic d’Oftalmologia (ICOF). Hospital Clínic de Barcelona.
CORRESPONDENCIA
Mª Socorro Alforja Castiella
E-mail: alforja@clinic.ub.es
E-mail: alforja@clinic.ub.es
Introducción
Desde que se publicaron los primeros artículos sobre tomografía de coherencia óptica (OCT, Optical Coherence Tomography) en fondos de ojos humanos in vitro, a principios de los años 1990, se ha producido un espectacular avance tecnológico que ha permitido evolucionar desde las iniciales Time Domain-OCT hasta las más recientes Ultrahigh Resolution-OCT (UHR-OCT) y Swept-Source OCT (SS-OCT). Con estas últimas, además de evaluar la retina de los pacientes con distrofias retinianas, puede evaluarse el estado de la coroides y posiblemente en un futuro correlacionarlo con el estado anatómico y funcional de la retina1. Es una prueba fácil de realizar, no invasiva, que no requiere contacto con la superficie ocular2 y que ayuda en la monitorización, tanto de la progresión de la enfermedad como de la respuesta a los tratamientos.Este avance en la tecnología de obtención de imágenes ha proporcionado una mejora en la calidad de las imágenes tomográficas que, asociada a un mejor conocimiento de la anatomía y la fisiología de la retina, ha permitido consensuar a un panel de expertos la nomenclatura para designar las bandas, las capas y las zonas retinianas con diferentes reflectividades observadas en la OCT, y correlacionarla con las estructuras anatómicas de la retina. De esta manera se distinguen cuatro bandas hiperreflectivas diferenciadas en la retina externa3,4:
- La más interna correspondería a la membrana limitante externa (MLE), la zona de unión de los segmentos internos de los fotorreceptores (FR) con las células de Muller.
- Inmediatamente por debajo se encuentra la zona de los elipsoides (EZ) de los segmentos internos de los FR.
- La tercera banda correspondería a la zona de interdigitación (IZ) de los segmentos externos de los FR con las células del epitelio pigmentario de la retina (EPR).
- Y por último, la más externa, es la línea correspondiente al complejo EPR-membrana de Bruch (MB).
OCT en voluntarios sanos
En la imagen superior de la Figura 1 puede verse una OCT macular realizada con HD- OCT (Cirrus de Zeiss) aumentada, y en la imagen inferior realizada con OCT de última generación DRI-OCT (Atlantis de Topcon).
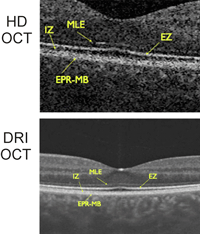
Figura 1. OCT en voluntarios sanos. Imagen superior: OCT realizada con HD-OCT (Cirrus de Zeiss); imagen inferior: OCT realizada con tecnología DRI-OCT (Atlantis de Topcon). En ambas imágenes pueden distinguirse las cuatro líneas hiperreflectivas de la retina externa (desde la parte vítrea o más interna a la parte coroidea o más externa): membrana limitante externa (MLE), zona de los elipsoides (EZ), zona de interdigitación (IZ) y complejo EPR-membrana de Bruch.

La presencia y la integridad de la EZ se han asociado a una mejor agudeza visual y grosor foveal en pacientes con RP, y pueden utilizarse, junto con la evaluación del anillo de autofluorescencia, como parámetros importantes en la monitorización de estos pacientes2,5. La longitud de la EZ se ha correlacionado con el área de campo visual5.
OCT patológica
En la Figura 2 se muestra un paciente con síndrome de Usher: en el recuadro verde (imagen superior derecha), la fóvea presenta una anatomía conservada, pero inmediatamente fuera de ella, en el recuadro rojo, se observa una pérdida de la anatomía normal a expensas de las cuatro capas externas, con atrofia del EPR. En la parte inferior de la composición se ofrece una imagen completa de la DRI-OCT.
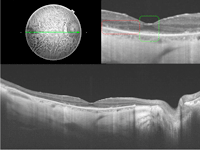
Figura 2. DRI-OCT patológica. Paciente con síndrome de Usher. Imagen superior izquierda: retinografía de fondo de ojo del paciente. Imagen superior derecha: DRI-OCT foveal; en el recuadro verde, ampliación de la fóvea, con anatomía conservada; inmediatamente fuera de ella, en el recuadro rojo, pérdida de la anatomía normal a expensas de las cuatro capas externas con atrofia del EPR. Imagen inferior de la composición: DRI-OCT completa en la que se ven tanto el nervio óptico como la mácula.

Fijándose en el espesor de la coroides en la imagen de DRI-OCT de este paciente (Figura 2, imagen inferior) y comparándola con la siguiente imagen de un voluntario sano (Figura 3), puede observarse una disminución en el espesor.
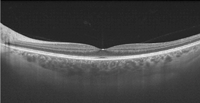
Figura 3. DRI-OCT macular en voluntario sano: puede observarse un mayor espesor de la coroides comparando con la DRI-OCT del paciente con distrofia retiniana de la Figura 2.
Por otro lado, no hay que olvidar que la OCT, al igual que otras pruebas complementarias, no debe valorarse de forma aislada sino en el contexto de cada paciente. En ocasiones, la falta de resolución o una señal pobre hacen difícil llegar a una conclusión3; por esto, será el conjunto de la anamnesis, la exploración ocular y las pruebas complementarias (pruebas electrofisiológicas, pruebas genéticas, OCT, autofluorescencia…) lo que ayude en el diagnóstico y el seguimiento de los pacientes.
Utilidad de la OCT en las distrofias retinianas
La OCT proporciona imágenes detalladas que permiten realizar un análisis capa por capa de las estructuras retinianas, lo que ha hecho que se convierta en una herramienta imprescindible en el día a día de las consultas6.Resulta de gran utilidad en:
- Ayuda en el diagnóstico y el seguimiento-progresión-pronóstico de la enfermedad7.
- Valoración de anomalías concomitantes vítreas, vitreoretinianas e intraretinianas.
- Valoración de la respuesta a los tratamientos.
Teniendo en cuenta la enorme variedad de distrofias que afectan a la retina/coroides, la gran diversidad fenotípica interpersonal, incluso intrafamiliar, y que son patologías muchas de ellas muy poco frecuentes, resulta difícil recopilar todas las alteraciones que pueden encontrarse en la OCT. A continuación se mencionan algunos de los posibles hallazgos y ejemplos de varios de ellos:
- Hialoisis asteroidea y otras condensaciones vítreas.
- Engrosamiento de la membrana limitante interna y de la membrana epiretiniana (MER).
- Tracción vitreomacular.
- Agujero macular/agujero lamelar.
- Edema macular cistoideo (EMQ).
- Esquisis retiniana.
- Desestructuración de las capas externas retinianas: cavitación, tubulaciones retinianas externas (TRE), atrofia foveal.
- Membranas neovasculares.
- Depósito de material patológico subretiniano.
- Otros hallazgos.
Hialosis asteroidea
Paciente con retinosis pigmentaria que asocia hialosis asteroidea: se aprecian cuerpos hiperreflectivos dispersos en la cavidad vítrea (Figura 4).

Figura 4. Paciente con retinosis pigmentaria que además asocia hialosis asteroidea: se aprecian cuerpos hiperreflectivos dispersos en la cavidad vítrea.

Membrana epiretiniana
Membrana epiretiniana en un paciente con retinosis pigmentaria: puede observarse como una línea hiperreflectiva sobre la superficie interna de la retina. En este paciente se observan además varios quistes intraretinianos que se aprecian como zonas redondeadas hiporreflectivas en el espesor de la retina (Figura 5).

Figura 5. Membrana epiretiniana en paciente con retinosis pigmentaria; puede observarse como una línea hiperreflectiva sobre la superficie interna de la retina. En este paciente se observan además varios quistes intraretinianos que se aprecian como zonas redondeadas hiporreflectivas en el espesor de la retina.

Síndrome de tracción vitreomacular
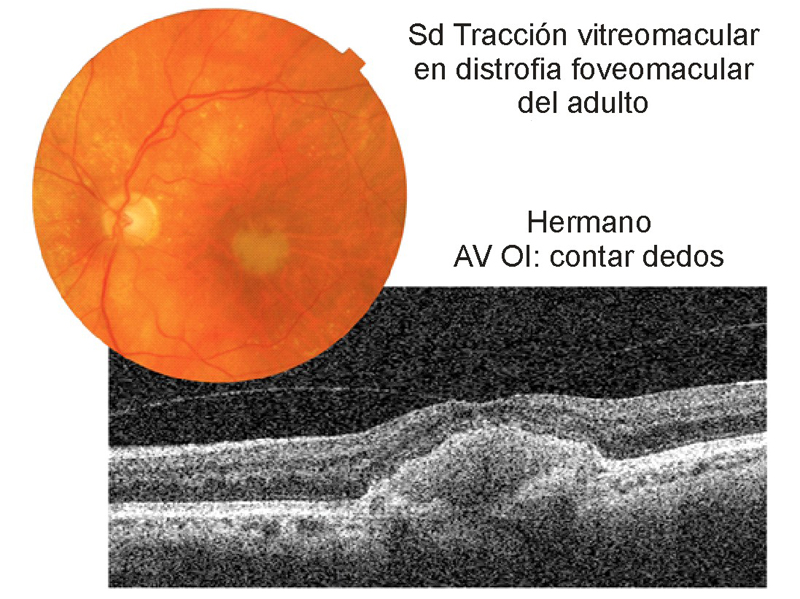
Dos hermanos, hombre de 92 años de edad (Figura 6 y Figura 7) y mujer de 86 años de edad (Figura 8, Figura 9 y Figura 10) con distrofia viteliforme del adulto que además asociaban en su ojo izquierdo un síndrome de tracción vitreomacular.
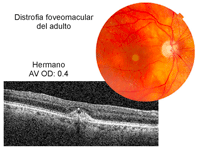
Figura 6. Distrofia viteliforme, foveomacular o pseudoviteliforme del adulto. Retinografía y OCT del ojo derecho de un varón de 92 años de edad. En la retinografía, lesión redonda amarillenta foveal que corresponde al depósito de lipofuscina. En la imagen tomográfica, elevación hiperreflectiva subfoveal correspondiente a la lipofuscina. En el ápex de la elevación, punto hiperreflectivo.

Figura 7. Distrofia viteliforme, foveomacular o pseudoviteliforme del adulto. Ojo izquierdo del paciente de la Figura 6. En la retinografía, lesión redondeada amarillenta, de mayor tamaño que la del ojo derecho. En la imagen tomográfica se ve claramente la adhesión del vítreo a la fóvea, con cierta tracción del perfil foveal. En este caso la lesión viteliforme es de mayor tamaño que en el ojo contralateral, y de contorno algo irregular.

En la OCT del ojo derecho (hombre, Figura 6) se observa material hiperreflectivo (lipofuscina) situado entre el EPR/MB y la retina neurosensorial9. En el ápex de la lesión se aprecia un punto hiperreflectivo, que se ha relacionado con expansión de la hiperreflectividad desde el EPR y los FR hacia la capa nuclear externa10.
Aunque habitualmente mantienen buenas agudezas visuales durante el curso de la enfermedad, finalmente la mayor parte de los pacientes desarrollan un deterioro10.
En el ojo izquierdo del mismo paciente (Figura 7) se aprecia una lesión de mayor tamaño, de superficie algo irregular y adhesión vítrea, que produce cierta tracción en la fóvea.
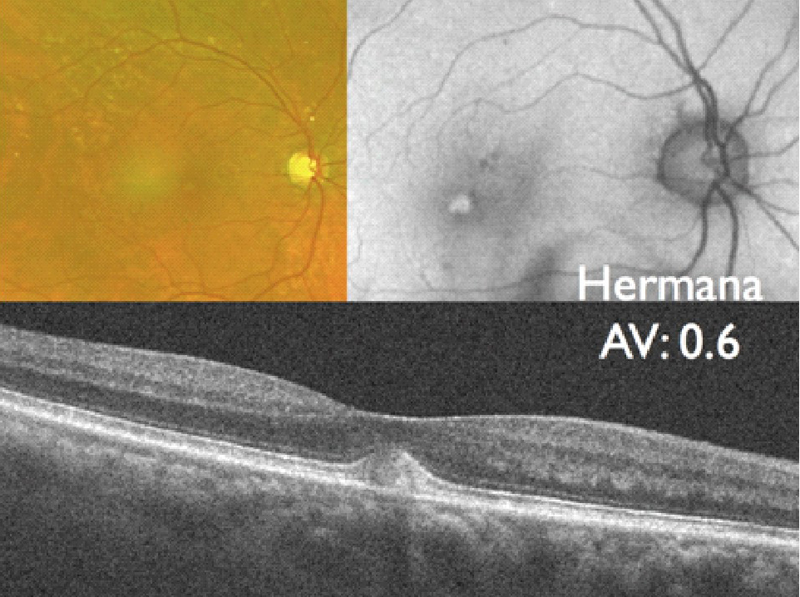
La hermana del paciente anterior presentaba en su ojo derecho (Figura 8) una lesión viteliforme hiperreflectiva homogénea y mantenía una agudeza visual de 0,6. En el ojo izquierdo (Figura 9) se observa una lesión viteliforme con presencia de nivel que se pone de manifiesto claramente en la OCT (Figura 10).
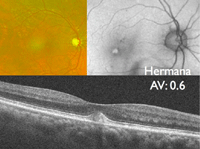
Figura 8. Distrofia viteliforme, foveomacular o pseudoviteliforme del adulto. Retinografía (imagen superior izquierda), autofluorescencia macular (imagen superior derecha) y OCT (imagen inferior) del ojo derecho de la hermana, de 86 años de edad, del paciente de las Figuras 6 y 7. En la autofluorescencia la lesión viteliforme se pone de manifiesto como una zona redondeada de hiperautofluorescencia en el centro de la mácula.

Figura 9. Distrofia viteliforme, foveomacular o pseudoviteliforme del adulto. Retinografía del ojo izquierdo de la paciente de la Figura 8. La lesión viteliforme presenta un nivel por acumulación de la lipofuscina en su parte inferior, de aspecto más opaco y amarillento que la parte superior de la lesión.

Figura 10. Distrofia viteliforme, foveomacular o pseudoviteliforme del adulto. Imagen de autofluorescencia de fondo y cortes tomográficos del ojo izquierdo de la paciente de las Figuras 8 y 9. El corte por la parte superior de la lesión (línea amarilla) corresponde a la zona hipoautofluorescente de la autofluorescencia y se traduce en la OCT como una lesión hiporreflectiva, ópticamente vacía. El corte por la parte inferior de la lesión (línea roja) se ve en la autofluorescencia como una zona hiperautofluorescente, y en la OCT como una elevación hiperreflectiva. Además puede verse, al igual que presenta su hermano, una adhesión vitreomacular foveal que ocasiona cierta tracción de la superficie foveal.


En la Figura 10, en el corte situado en la parte superior de la lesión (amarillo), puede verse un espacio ópticamente vacío que también se aprecia en la imagen de autofluorescencia (AF) como una zona hipoautofluorescente. En el corte por la parte inferior de la lesión (rojo) puede verse la lesión homogénea y la adhesión vitreomacular que provoca una configuración en meseta de la retina.
En el siguiente ejemplo (Figura 11, Figura 12 y Figura 13), paciente afecta de retinosis pigmentaria. En las imágenes de OCT (Figura 12 y Figura 13) puede verse una hialoides claramente engrosada y unida focalmente a la retina en algunos puntos extrafoveales (flecha blanca) (Figura 12) y desprendida en otras (flecha blanca) (Figura 13).

Figura 11. Paciente con retinosis pigmentaria. Retinografías (imágenes superiores) y autofluorescencia (imágenes inferiores) de campo amplio con Optomap.

Figura 12.

Figura 13.
Figuras 12 y 13. Paciente con retinosis pigmentaria. OCT del mismo paciente realizada en distintos puntos de la mácula, con evidencia de zonas de adhesión (Figura 12) y zonas de desprendimiento vítreo (Figura 13). En la OCT de la Figuras 12 y 13 puede apreciarse el engrosamiento de la hialoides.



Maculopatía quística: edema macular cistoideo y pseudoquistes
Algunos pacientes con distrofias retinianas presentan en la OCT lesiones quísticas en la retina. Cuando en la angiografía fluoresceínica se observa nula o mínima acumulación de contraste se habla de pseudoquistes (non-CME macular cysts), que se forman en determinadas distrofias por una alteración en la anatomía normal de la retina en la mácula, como es el caso de la retinosquisis ligada al X11.Se encuentran cavidades quísticas en la OCT de diversas distrofias12,13: RP, retinosquisis ligada al X, retinopatía NR2E3 (enhaced S-cone syndrome, enfermedad de Goldmann-Favre y distrofia retiniana con pigmento agrupado [clumped pigmentari retinal dystrophy]), coroideremia, atrofia gyrata y drusas familiares.
En el siguiente caso (Figura 14 y Figura 15) se presenta una paciente con síndrome de Usher que asocia grandes cavidades quísticas en ambos ojos, que mejoran parcialmente con inhibidores de la anhidrasa carbónica por vía oral.

Figura 14. Retinosis pigmentaria en un paciente con síndrome de Usher. Ojo derecho, imagen superior izquierda, retinografía de campo amplio; imagen superior derecha, autofluorescencia de campo amplio; imagen inferior, OCT que muestra grandes cavidades quísticas intraretinianas que pueden verse como zonas redondeadas u ovoides que ocupan prácticamente la totalidad del espesor retiniano en su zona más central, y otras más pequeñas que afectan las capas externas retinianas en la zona perifoveal.

Figura 15. Retinosis pigmentaria en un paciente con síndrome de Usher. Ojo izquierdo, imagen superior izquierda, retinografía de campo amplio; imagen superior derecha, autofluorescencia de campo amplio; imagen inferior, OCT con cavidades quísticas que afectan preferentemente a las capas externas.


Como ejemplo de cavidades quísticas, en las Figuras 16-21 se muestran dos hermanos varones, de 40 y 45 años de edad, afectos de retinosquisis familiar ligada al X:
- Varón de 40 años: atrofia central macular bilateral (Figura 16: retinografías, parte superior) y mínima hiperfluorescencia desde tiempos iniciales de la angiografía, que no varía a lo largo del angiograma (Figura 16: angiografía, parte inferior). En la imagen de OCT (Figura 17) se ven grandes cavidades quísticas con pérdida de la arquitectura normal foveal.
- Varón de 45 años (Figura 18 y Figura 19): a pesar de tener espacios quísticos maculares escasos en ambos ojos, presentaba una pérdida de la estructura normal de las cuatro bandas en la retina externa, con agudezas visuales de 0,2 en el ojo derecho y 0,1 en el izquierdo.

Figura 16. Retinosquisis familiar ligada al X. Varón de 40 años. Imágenes superiores, retinografías de campo amplio en las que se observa atrofia central macular bilateral. Imágenes inferiores, angiografía fluoresceínica de campo amplio que muestra leve hiperfluorescencia macular desde tiempos iniciales, que no varía a lo largo del angiograma.

Figura 17. Retinosquisis familiar ligada al X. Imagen de OCT del paciente de la Figura 16. La OCT superior corresponde al ojo derecho y la OCT inferior corresponde al ojo izquierdo. En ambos ojos se ven grandes cavidades quísticas que ocupan el espesor completo retiniano, con pérdida de la arquitectura normal foveal.

Figura 18.

Figura 19.
Figuras 18 y 19. Retinosquisis familiar ligada al X. Ojo derecho (imágenes superiores) y ojo izquierdo (imágenes inferiores) de paciente varón de 45 años de edad, hermano del paciente de las Figuras 16 y 17. En la OCT de ambos ojos se aprecian pequeños espacios quísticos maculares intraretinianos, pero con importante desestructuración de las capas externas de la retina en la zona de la fóvea.
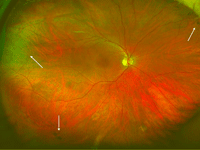
Figura 20.

Figura 21.
Figuras 20 y 21. Retinosquisis familiar ligada al X. Retinografías de campo amplio del ojo derecho (Figura 20) y del ojo izquierdo (Figura 21) del hermano de 45 años de edad. En la retina periférica temporal del ojo derecho (flecha blanca superior), retinosquisis temporal, y tanto en la periferia nasal del ojo derecho como en la temporal de ambos ojos se observan algunos acúmulos de pigmento numular (flechas blancas).






En la retina periférica de ambos pacientes podía apreciarse en la exploración detallada del fondo de ojo una retinosquisis temporal (Figura 20: retinografía del ojo derecho del hermano mayor) y algunos acúmulos de pigmento numular (Figura 20 y Figura 21: retinografía del ojo izquierdo del hermano mayor).
En el caso concreto de los pacientes con RP, el deterioro progresivo visual afecta fundamentalmente a la visión nocturna y la visión periférica, pero puede verse agravado por patologías asociadas de la interfase vitreoretiniana. Debido a la elevada incidencia de EMQ, la realización de OCT se hace necesaria para el correcto seguimiento de estos pacientes5. Es posible que el edema macular de las distrofias retinianas tenga una causa multifactorial: de un lado, la muerte celular (FR); de otro, una respuesta inflamatoria secundaria a la destrucción de fotorreceptores. También se ha implicado una disfunción en el EPR y, asociado a los anteriores, un componente traccional relacionado con alteraciones en la interfase vitreoretiniana6.
En la evolución de algunos pacientes con edema macular puede producirse un agujero macular lamelar, como en la siguiente paciente con retinosis pigmentaria, que inicialmente desarrolló edema sólo en el ojo izquierdo (Figura 23, inferior izquierda) y varios años después en el ojo derecho (Figura 22). El ojo izquierdo progresó hacia la formación de un agujero lamelar macular (Figura 23, inferior derecha).
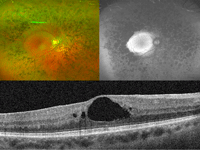
Figura 22.

Figura 23.
Figuras 22 y 23. Retinosis pigmentaria con edema macular cistoideo en OCT de ojo derecho (Figura 22, inferior), y en ojo izquierdo edema macular (Figura 23, inferior izquierda) que evoluciona a agujero macular lamelar (Figura 23, inferior derecha).


Cavitación foveal
El término cavitación foveal3,6,14-17 hace referencia a la pérdida focal de la reflectividad foveal externa en las imágenes de OCT. Este espacio hiporreflectivo puede verse en la enfermedad de Stargardt, la acromatopsia congénita y en todas aquellas distrofias en las que exista una disfunción de conos.Este hallazgo se cree debido a la pérdida de los segmentos externos de los conos, manteniéndose íntegros los bastones, más largos, y que harían de soporte estructural. Se considera además un hallazgo precoz de la enfermedad, puesto que conforme avanza el daño retiniano se produce un colapso del mismo y dejaríamos de visualizarlo.
En las imágenes de DRI-OCT (Figura 24 y Figura 25) puede verse la cavitación foveal (flecha roja en ambos ojos) en un paciente con distrofia de conos. La gran definición de las imágenes permite visualizar además otros detalles, como la cavidad vítrea premacular en el ojo izquierdo (Figura 25, asterisco rojo).

Figura 24. En la imagen inferior, DRI-OCT del ojo derecho de un paciente afecto de distrofia de conos. La imagen superior muestra la OCT a más aumento, en la que se aprecia la cavitación foveal como una zona hiporreflectiva en las capas externas, subfoveal.

Figura 25. En la imagen inferior, la DRI-OCT del ojo izquierdo del paciente anterior muestra una cavidad vítrea premacular (asterisco rojo), y en la imagen superior, a más aumento, se aprecia la cavitación foveal.


En una paciente de 32 años de edad (Figura 26, Figura 27 y Figura 28), con mácula en ojo de buey secundaria a distrofia de conos, puede observarse también el mismo hallazgo (Figura 26). En las siguientes imágenes se muestran las retinografías (Figura 27, superior), la imagen de OCT Cirrus (Figura 27, inferior) y la imagen de DRI-OCT (Figura 28). Al interrogar específicamente a la paciente por los antecedentes oculares en la familia, explica baja visión en su madre (MAVC: 0,05 AO), de causa no filiada. A la exploración la madre presentaba estafiloma macular llamativo bilateral, que puede apreciarse con detalle en las retinografías con Optomap (Figura 29 y Figura 30), la autofluorescencia (Figura 29 y Figura 30) y la DRI-OCT (Figura 31 y Figura 32).

Figuras 26. Paciente de 32 años de edad con mácula en ojo de buey secundaria a distrofia de conos. OCT con cavitación foveal en ambos ojos.

Figura 27. Paciente de 32 años de edad con mácula en ojo de buey secundaria a distrofia de conos. Retinografías en las que se aprecia la mácula en ojo de buey en ambos ojos.
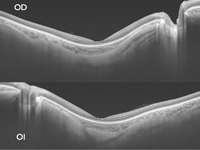
Figura 28. Paciente de 32 años de edad con mácula en ojo de buey secundaria a distrofia de conos. DRI-OCT que pone de manifiesto el estafiloma posterior en ambos ojos.
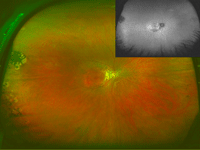
Figura 29.
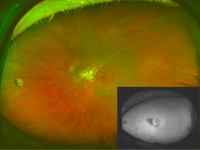
Figura 30.

Figura 31.
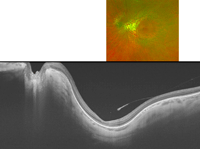
Figura 32.
Figuras 29 a 32. Distrofia de conos asociada a estafiloma posterior en la madre de la paciente de las Figuras 26 a 28. Retinografía y autofluorescencia de campo amplio que muestran atrofia peripapilar y estafiloma posterior en ojo derecho y en ojo izquierdo (Figura 30). Figuras 31 y 32: imagen de DRI-OCT de ambos ojos, en la que se aprecia el estafiloma posterior llamativo en los dos ojos.







Tubulaciones retinianas externas
En la OCT se ven las tubulaciones retinianas externas18,19 como estructuras redondeadas u ovoides de pared hiperreflectiva e interior hiporreflectivo (Figura 33, Figura 34 y Figura 35, flecha roja) que aparecen en relación a zonas de atrofia retiniana (Figura 35, línea amarilla) de distinta etiología (degeneración macular asociada a la edad, distrofias retinianas, coroidopatía serosa central crónica), en las cuales han sido dañados los FR y el EPR. Se considera un hallazgo inespecífico y propio de estadios avanzados. Desde el punto de vista anatomopatológico, parece que las TRE se desarrollan por una progresiva reorganización e invaginación de los FR, rodeados de MLE. Se desconoce hasta el momento el mecanismo patogénico que lleva a la formación de tales estructuras.

Figura 33. Señalada con una flecha roja, tubulación retiniana externa: estructura redonda u ovalada de pared hiperreflectiva rodeando una zona hiporreflectiva.

Figura 34. Tubulación retiniana externa adyacente a una zona de importante atrofia foveal.

Figura 35. Tubulación retiniana externa, señalada con una flecha roja, al lado de una zona de atrofia retiniana, señalada con línea amarilla.



Atrofia macular
La atrofia macular aparece como el estadio final de múltiples distrofias retinianas. Como ejemplo se presenta un paciente afecto de distrofia coroidea areolar central, con atrofia grave en el polo posterior, que permite la visualización de los grandes vasos de la coroides (Figura 36: retinografía; Figura 37: autofluorescencia; Figura 38: OCT).

Figura 36. Distrofia coroidea areolar central. Retinografías de campo amplio de ambos ojos en las que se aprecia la atrofia macular bilateral.
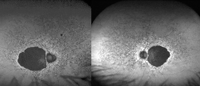
Figura 37. Distrofia coroidea areolar central. Imagen de autofluorescencia con hipoautofluorescencia macular bilateral con patrón de hiper-hipoautofluorescencia moteada en el resto de la retina de ambos ojos.

Figura 38. Distrofia coroidea areolar central. OCT: atrofia macular grave con disminución del espesor retiniano y aumento de la hiperreflectividad por debajo de la retina, debido a la atrofia retiniana.



En la OCT (Figura 38) puede observarse la disminución del espesor de todas las capas de la retina neurosensorial con ausencia del EPR y un aumento de la reflectividad posterior debido al mayor paso de luz a través de la retina atrófica.
Depósito de material subretiniano
Las distrofias en patrón engloban un grupo de enfermedades del EPR en las que se acumula material extracelular procedente de los FR y del EPR bajo la retina neurosensorial9,10,20-22. Entre ellas se distinguen la distrofia foveomacular del adulto o viteliforme del adulto, la distrofia en alas de mariposa, la distrofia en patrón multifocal, la distrofia reticular de Sjögren y, por último, la distrofia fundus pulverulentus.Como ejemplo se presenta una paciente de 83 años de edad con distrofia viteliforme del adulto (Figuras 39-42). Aunque la progresión resulta evidente en las retinografías realizadas (Figura 39), la OCT aporta mayor información sobre las estructuras retinianas (Figura 40 y Figura 41). En las retinografías, además, se aprecia un nevus coroideo (Figura 39, derecha).
En las imágenes de OCT de ambos ojos vemos la progresión de la enfermedad a lo largo de los últimos 6 años: en el ojo derecho (Figura 40) se ve el material viteliforme homogéneo, y que con la evolución se va produciendo la reabsorción de este material para finalmente dejar una zona de atrofia geográfica.
Al diagnóstico, el ojo izquierdo (Figura 41) se encontraba en fase más avanzada. Puede apreciarse una TRE en el borde izquierdo de la atrofia en el estadio final de la enfermedad.
En las imágenes de AF (Figura 42) se observa que la lesión no es sólo foveal, sino que muestra un patrón reticular en ambas arcadas superiores y la zona temporal macular.

Figura 39. Distrofia viteliforme del adulto. Retinografías que muestran la evolución del proceso entre las imágenes iniciales (superiores) y las imágenes tras años de seguimiento. En las imágenes superiores se aprecia el depósito viteliforme foveal en ambos ojos con pigmento, y en las imágenes inferiores se aprecia la atrofia foveal que deja ver los grandes vasos de la coroides. Además, se observa un nevus coroideo en el polo posterior del ojo izquierdo que no se modifica tras años de seguimiento.

Figura 40. Distrofia viteliforme del adulto. Ojo derecho. A) Material viteliforme subfoveal homogéneo. B) Reabsorción parcial del material viteliforme. C) Atrofia geográfica tras la reabsorción completa del material.

Figura 41. Distrofia viteliforme del adulto. Ojo izquierdo. A) En el momento del diagnóstico, el ojo izquierdo se encontraba en una fase más avanzada que el derecho, mostrando reabsorción parcial del material viteliforme. B) Estructura tubular retiniana externa (TRE) en el borde izquierdo de la atrofia en el estadio final de la enfermedad. Atrofia macular grave que condiciona la hiperreflectividad por debajo de la fóvea.

Figura 42. Distrofia viteliforme del adulto. En el mismo paciente, con autofluorescencia de campo amplio se observa que las alteraciones no son sólo foveales, sino que muestra un patrón reticular en ambas arcadas superiores y la zona temporal macular. En ocasiones, estas alteraciones pueden pasar desapercibidas en la exploración del fondo de ojo.




El siguiente paciente (Figura 43, Figura 44 y Figura 45) presenta una distrofia en patrón reticular en el contexto de una glucogenosis tipo V o enfermedad de McArdle. En las imágenes de fondo de ojo (Figura 43, superior) puede verse el material con patrón lineal irregular de coloración amarillenta en la mácula de ambos ojos. La imagen de OCT muestra un estadio más avanzado en el ojo izquierdo (Figura 43, inferior derecha) con importante atrofia de capas externas; el ojo derecho presenta depósito subretiniano subfoveal (Figura 43, inferior izquierda).
Del mismo modo que en el caso de la distrofia viteliforme del adulto, en las siguientes imágenes de autofluorescencia (Figura 44: ojo derecho; Figura 45: ojo izquierdo) se ponen de manifiesto más lesiones con patrón reticular fuera del área macular en ambos ojos.
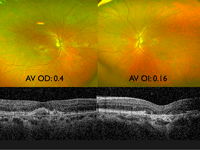
Figura 43. Distrofia en patrón reticular en un paciente con glucogenosis tipo V o enfermedad de McArdle. Imágenes superiores, retinografías de campo amplio: se aprecia depósito de material amarillento perifoveal siguiendo un patrón radial perifoveal y lineal irregular en la retina nasal. Imagen inferior izquierda, OCT de ojo derecho que presenta depósito subretiniano subfoveal. Imagen inferior derecha, OCT de ojo izquierdo que en el momento del diagnóstico presenta una fase más avanzada, con importante atrofia de las capas externas.

Figura 44.
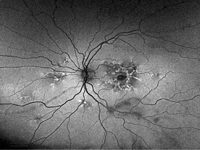
Figura 45.
Figuras 44 y 45. Distrofia en patrón reticular en un paciente con glucogenosis tipo V o enfermedad de McArdle. La autofluorescencia (Figura 44, ojo derecho; Figura 45, ojo izquierdo) muestra un patrón reticular hiperautofluorescente perifoveal y fuera del área macular, sobre todo retina nasal en ambos ojos.



Otros hallazgos
Aunque de forma infrecuente, está descrita en la literatura médica la asociación de RP y coroidopatía serosa central (CSC)23.Se presenta el caso de un varón de 32 años de edad, originario de Pakistán, con retinosis pigmentaria asociada a MER bilateral y CSC en el ojo izquierdo (Figura 46 y Figura 47). En la primera visita (Figura 47) su mejor agudeza visual corregida (MAVC) en el ojo izquierdo era de 0,2, a los 3 meses era de 0,4 y a los 6 meses era de 0,7, con resolución espontánea del cuadro.
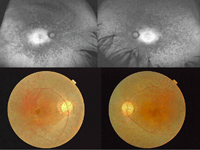
Figura 46. Varón de 32 años de edad, originario de Pakistán, con retinosis pigmentaria asociada a MER bilateral y CSC en el ojo izquierdo. Imágenes superiores, autofluorescencia; imágenes inferiores, retinografías.
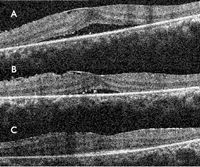
Figura 47. OCT de ojo izquierdo del paciente de la Figura 46. A) Fluido subretiniano subfoveal al diagnóstico. B) A los 3 meses, disminución del fluido. C) A los 6 meses, resolución total del fluido.


La OCT en la valoración de la respuesta a los tratamientos
Aunque la vitrectomía y algunos tratamientos intravítreos han demostrado una mejora anatómica de la retina, no siempre esta se correlaciona con una mejora en la función visual del paciente, ya que puede estar limitada por la disfunción retiniana de larga evolución; de aquí se deduce la necesidad de valorar detenidamente los pros y los contras de los distintos tratamientos e informar al paciente de forma detallada sobre los mismos antes de realizarlos.En la Figura 48 se muestra un paciente con edema macular cistoideo en el ojo derecho en un contexto de retinosis pigmentaria ligada al X. Se aprecian los espacios quísticos intraretinianos con una MAVC de 0,2, y una mejora anatómica del edema tras 1 mes de tratamiento con acetazolamida por vía oral. Su MAVC pasó a ser de 0,4.
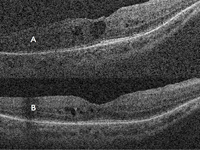
Figura 48. Retinosis pigmentaria ligada al X. Edema macular cistoideo en ojo derecho. A) Espacios quísticos intraretinianos. B) Mejora anatómica del edema tras 1 mes de tratamiento con acetazolamida por vía oral.

El siguiente paciente (Figura 49), afecto de retinosis pigmentaria de herencia recesiva no asociada a otras patologías sistémicas, presentaba llamativa sintomatología de visión borrosa, miodesopsias y fotofobia en su ojo derecho. En la exploración presentaba en el ojo derecho hialosis asteroidea (Figura 49: retinografía y OCT, superior) con una MAVC de 0,6, frente al ojo izquierdo con MAVC de 0,9.

Figura 49. Retinosis pigmentaria en ojo derecho asociada a hialosis asteroidea. Retinografía y OCT. A) Previtrectomía. B) Posvitrectomía.

Se le realizó una vitrectomía y se obtuvo un resultado satisfactorio por parte del paciente, ya que mejoró subjetivamente la sensación de borrosidad (Figura 49: OCT, inferior). A pesar de esto, no se encontró cambio en la MAVC tras la cirugía.
La OCT se presenta como una herramienta eficaz en la valoración del estado macular, incluso en pacientes con hialosis asteroidea llamativa, en los que la exploración detallada del fondo de ojo puede resultar dificultosa.
En los pacientes con distrofias que asocian MNVSR, el uso de la OCT se hace imprescindible para evaluar la progresión o la estabilidad de las lesiones tras el tratamiento intravítreo.
En la Figura 50, Figura 51, Figura 52 y Figura 53 se muestra un paciente con distrofia en patrón reticular asimétrica que presentaba metamorfopsia asociada a neovascularización subretiniana en el ojo izquierdo, y que fue tratado con tres inyecciones de ranibizumab intravítreo. Después de 3 años de seguimiento sin más tratamiento, el paciente se mantiene estable sin nuevas recidivas.

Figura 50. Distrofia en patrón reticular asimétrica. Retinografías: en el ojo derecho se aprecia atrofia peripapilar, y en ojo izquierdo atrofia peripapilar asociada a depósito de material blanco amarillento radial perifoveal.

Figura 51. Distrofia en patrón reticular asimétrica. Autofluorescencia: ojo derecho, hipoautofluorescencia peripapilar con pequeño halo de hiperautofluorescencia; ojo izquierdo, hipoautofluorescencia peripapilar con líneas hiperautofluorescentes en patrón radial desde la fóvea.

Figura 52. Distrofia en patrón reticular asimétrica. Imágenes superiores, hiperfluorescencia central puntiforme correspondiente a neovascularización subretiniana en el ojo izquierdo. Imágenes inferiores, OCT: en la imagen inferior derecha, el depósito de material subretiniano asociado a neovascularización subretiniana.

Figura 53. Distrofia en patrón reticular asimétrica. A) Antes del tratamiento con ranibizumab. B) Un año después del tratamiento. C) Tres años después del tratamiento.




Los dos pacientes presentados a continuación (Figuras 54- 57) son hermanos, varones de 35 y 40 años de edad, con sospecha de distrofia pseudoinflamatoria de Sorsby. El padre, ya fallecido, presentaba el mismo cuadro que sus dos hijos. Ambos requieren un control estricto, con inyecciones repetidas de antiangiogénicos, muy similar al de los pacientes afectos de degeneración macular asociada a la edad por formación de membranas neovasculares. Debido al importante componente fibrótico de las lesiones, con la OCT podemos visualizar pequeños cambios retinianos y nuevas recidivas, que en muchos casos escapan a la exploración del fondo de ojo.
- Hermano de 35 años (Figura 54: retinografía, superior, y AF inferior; Figura 55: OCT). En la siguiente imagen de OCT: ojo derecho (Figura 55, superior), relativa conservación de la fóvea; en la OCT del ojo izquierdo (Figura 55, inferior), importante lesión cicatricial y atrofia de la retina neurosensorial en la fóvea.
- Hermano de 40 años (Figura 56: retinografía, superior, y AF inferior; Figura 57). En la imagen de OCT (Figura 57: ojo derecho, superior; ojo izquierdo, inferior) puede apreciarse el importante componente fibrótico de las lesiones.

Figura 54. Distrofia pseudoinflamatoria de Sorsby. Hermano de 35 años de edad. En las retinografías superiores se aprecia afectación bilateral en un estadio más evolucionado y fibrótico en el ojo izquierdo. AF inferiores: patrón macular con áreas de hiperautofluorescencia e hipoautofluorescencia.
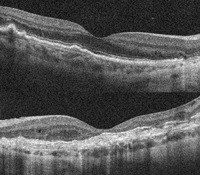
Figura 55. Distrofia pseudoinflamatoria de Sorsby. Hermano de 35 años de edad. OCT, superior, ojo derecho: relativa conservación de la fóvea; inferior, ojo izquierdo: importante lesión cicatricial y atrofia de la retina neurosensorial en la fóvea.

Figura 56. Distrofia pseudoinflamatoria de Sorsby. Hermano de 40 años de edad. Las retinografías, superior, muestran focos de atrofia y de fibrosis secundarios a MNVSR recidivantes, y la AF, inferior, muestra un patrón de hiperautofluorescencia e hipoautofluorescencia.

Figura 57. Distrofia pseudoinflamatoria de Sorsby. Hermano de 40 años de edad. OCT que muestra importante componente fibrótico de las lesiones tanto en el ojo derecho (superior) como en el ojo izquierdo (inferior).




De especial interés es también la utilización de la OCT en el seguimiento de los pacientes afectos de distrofias retinianas tras la cirugía de catarata. La siguiente paciente (Figura 58 y Figura 59), con retinosis pigmentaria, desarrolló edema macular tras ser intervenida de catarata. En las imágenes de OCT previas a la cirugía (Figura 59, superior) no presentaba edema macular. A los 15 días de la cirugía no complicada (Figura 59, inferior) presentaba quistes intraretinianos. Tras 1 mes con tratamiento tópico antiinflamatorio (antiinflamatorio no esteroideo y corticoides) no se obtuvo respuesta favorable, manteniendo el mismo edema que antes de comenzar el tratamiento médico.

Figura 58. Retinosis pigmentaria sectorial. Imágenes superiores, retinografías en las que se aprecia la afectación de la hemirretina inferior simétrica en ambos ojos. Imágenes inferiores, autofluorescencia: hipoautofluorescencia en la retina inferior con hiperautofluorescencia en anillo parcial inferior en ambos ojos.
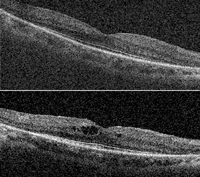
Figura 59. Retinosis pigmentaria sectorial. OCT, superior, previa a la cirugía de catarata en el ojo derecho; inferior, tras la cirugía de catarata mediante facoemulsificación, se observan quistes intraretinianos que no se modificaron a pesar del tratamiento (antiinflamatorio no esteroideo y esteroide tópico).


En conclusión, puede decirse que la OCT, desde sus orígenes con la Time Domain- OCT hasta el momento actual, con el desarrollo de nuevos dispositivos que permiten una mayor resolución de las imágenes retinianas, se ha mostrado como una herramienta eficaz para el diagnóstico y el seguimiento de la evolución y de los tratamientos en los pacientes con distrofias retinianas. No obstante, no hay que olvidar que la OCT es una prueba complementaria a la exploración del paciente y a otras pruebas que resultan necesarias para su correcto control.
Bibliografía
- Adhi M, Duker JS. Optical coherence tomography - current and future applications. Curr Opin Ophthalmol. 2013;24(3):213-21.
- Mitamura Y, Mitamura-Aizawa S, Nagasawa T, Katome T, Eguchi H, Naito T. Diagnostic imaging in patients with retinitis pigmentosa. J Med Invest. 2012;59(1-2):1-11.
- Lima LH, Sallum JM, Spaide RF Outer retina analysis by optical coherence tomography in cone-rod dystrophy patients. Retina. 2013;33(9):1877-80.
- Staurenghi G, Sadda S, Chakravarthy U, Spaide RF; International Nomenclature for Optical Coherence Tomography (IN•OCT) Panel*Proposed Lexicon for Anatomic Landmarks in Normal Posterior Segment Spectral-Domain Optical Coherence Tomography: The IN•OCT Consensus. Ophthalmology. 2014 Apr 19.
- Fischer MD, Fleischhauer JC, Gillies MC, Sutter FK, Helbig H, Barthelmes D. A new method to monitor visual field defects caused by photoreceptor degeneration by quantitative optical coherence tomography. Invest Ophthalmol Vis Sci. 2008;49(8):3617-21.
- Inui E, Oishi A, Oishi M, Ogino K, Makiyama Y, Gotoh N, et al. Tomographic comparison of cone-rod and rod-cone retinal dystrophies. Graefes Arch Clin Exp Ophthalmol. 2014;252(7):1065-9.
- Guía clínica para el diagnóstico diferencial y el manejo de las enfermedades hereditarias de la retina y coroides (primera revisión). Guías de Práctica clínica de la SERV (primera revisión: 2012)
- Triolo G, Pierro L, Parodi MB, De Benedetto U, Gagliardi M, Manitto MP, et al. Spectral domain optical coherence tomography findings in patients with retinitis pigmentosa. Ophthalmic Res. 2013;50(3):160-4.
- Finger RP, Charbel Issa P, Kellner U, Schmitz-Valckenberg S, Fleckenstein M, Scholl HP, et al. Spectral domain optical coherence tomography in adult-onset vitelliform macular dystrophy with cuticular drusen. Retina. 2010;30(9):1455-64.
- Hannan SR, de Salvo G, Stinghe A, Shawkat F, Lotery AJ. Common spectral domain OCT and electrophysiological findings in different pattern dystrophies. Br J Ophthalmol. 2013;97(5):605-10.
- Ganesh A, Stroh E, Manayath GJ, Al-Zuhaibi S, Levin AV. Macular cysts in retinal dystrophy. Curr Opin Ophthalmol. 2011;22(5):332-9.
- Yzer S, Barbazetto I, Allikmets R, van Schooneveld MJ, Bergen A, Tsang SH, et al. Expanded clinical spectrum of enhanced S-cone syndrome. JAMA Ophthalmol. 2013;131(10):1324-30.
- Querques G, Guigui B, Leveziel N, Querques L, Bandello F, Souied EH. Multimodal morphological and functional characterization of Malattia Leventinese. Graefes Arch Clin Exp Ophthalmol. 2013;251(3):705-14.
- Takahashi H, Hayashi T, Tsuneoka H, Nakano T, Yamada H, Katagiri S, et al. Occult macular dystrophy with bilateral chronic subfoveal serous retinal detachment associated with a novel RP1L1 mutation (p.S1199P). Doc Ophthalmol. 2014;129(1):49-56.
- Sisk RA, Leng T. Multimodal imaging and multifocal electroretinography demonstrate autosomal recessive stargardt disease may present like occult macular dystrophy. Retina. 2014;16.
- Zahlava J, Lestak J, Karel I. Optical coherence tomography in progressive cone dystrophy. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2013 Mar 21.
- Cho SC, Woo SJ, Park KH, Hwang JM. Morphologic characteristics of the outer retina in cone dystrophy on spectral-domain optical coherence tomography. Korean J Ophthalmol. 2013;27(1):19-27.
- Raja MS, Goldsmith C, Burton BJ. Outer retinal tubulations in maternally inherited diabetes and deafness (MIDD)-associated macular dystrophy. Graefes Arch Clin Exp Ophthalmol. 2013;251(9):2265-7.
- Goldberg NR, Greenberg JP, Laud K, Tsang S, Freund KB. Outer retinal tubulation in degenerative retinal disorders. Retina. 2013;33(9):1871-6.
- Zerbib J, Querques G, Massamba N, Puche N, Tilleul J, Lalloum F, et al. Reticular pattern dystrophy of the retina: a spectral-domain optical coherence tomography analysis. Am J Ophthalmol. 2013;156(6):1228-37.
- Puche N, Querques G, Benhamou N, Tick S, Mimoun G, Martinelli D, et al. High-resolution spectral domain optical coherence tomography features in adult onset foveomacular vitelliform dystrophy. Br J Ophthalmol. 2010;94(9):1190-6.
- Querques G, Regenbogen M, Quijano C, Delphin N, Soubrane G, Souied EH. High-definition optical coherence tomography features in vitelliform macular dystrophy. Am J Ophthalmol. 2008;146(4):501-50.
- Rootman DB, Mandelcorn E, Dracopoulos A, Boyd S, Mandelcorn MS. Concurrence of retinitis pigmentosa and central serous retinopathy. Digit J Ophthalmol. 2011;17(4):55-7.