2Unidad Oftalmología Pediátrica, Hospital Universitari Vall d’Hebron. Barcelona
3Servicio Oftalmología, Hospital Universitari Bellvitge. Barcelona
4Unidad Neurología Pediátrica, Hospital Universitari Vall d’Hebron. Barcelona
CORRESPONDENCIA
Servicio Oftalmología
Hospital Mútua de Terrassa. Barcelona
E-mail: saintgerons@gmail.com
Enfermedades neurodegenerativas
Las enfermedades neurodegenerativas son un conjunto de trastornos de origen genético que afectan de forma preferencial bien a la sustancia blanca o a la sustancia gris. Cuando la afectación predominante es de sustancia blanca se manifiestan como disfunción de la vía cortico-espinal, neuropatía periférica y atrofia óptica. Las enfermedades que afectan a la sustancia gris, por el contrario, se presentan con crisis comiciales, trastornos del movimiento y demencia. En muchas ocasiones la afectación es mixta, tanto de sustancia blanca y de gris, bien sea desde el inicio del proceso o en el curso evolutivo. En este apartado se revisan las enfermedades neurodegenerativas que debutan en la edad pediátrica y cursan con atrofia óptica. Se pueden distinguir dos grandes grupos (Tabla 1):- Los procesos con un defecto metabólico conocido y estos a su vez se han clasificado en errores congénitos del metabolismo intermediario y errores congénitos del metabolismo de las organelas celulares.
- Otras enfermedades neurodegenerativas genéticas con defecto metabólico desconocido.
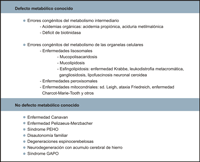
Tabla 1. Clasificación de las enfermedades neurodegenerativas.

Errores congénitos del metabolismo intermediario
Acidemias orgánicas
La acidemia propiónica y metilmalónica son enfermedades muy infrecuentes y de herencia autosómica recesiva. La clínica se suele iniciar en el periodo neonatal con alteraciones gastrointestinales inespecíficas. Posteriormente aparecen las manifestaciones neurológicas graves asociadas a cetoacidosis, hiperamoniemia y fallo multiorgánico.
Acidemia propiónica
- Base molecular: déficit de propionil-CoA carboxilasa.
- Clínica oftalmológica: los varones suelen desarrollar una atrofia óptica bilateral, es raro que la presenten las mujeres.
Aciduria metilmalónica
- Base molecular: déficit de metilmalonil CoA mutasa y/o adenosilcobalamina. Se trata de una enfermedad genéticamente heterogénea del metabolismo de la cobalamina y del metilmalonato. Se han descrito varios subgrupos (cb1A, cb1B, cb1C, cb1D y cb1F), los tres últimos se asocian con homocistinuria.
- Clínica oftalmológica: solo el subgrupo cb1C presenta manifestaciones oftalmológicas como maculopatía y atrofia óptica.
Déficit de biotinidasa
- Herencia: autosómica recesiva.
- Base molecular: mutación del gen biotinidasa (BTD) en el cromosoma 3p25.
- Clínica sistémica: la clínica suele aparecer durante los primeros meses de vida con retraso del desarrollo, convulsiones, sordera neurosensorial, paraparesia espástica, rash cutáneo, infecciones, ataxia aguda intermitente, alopecia y estridor.
- Manifestaciones oftalmológicas: atrofia óptica.
- Diagnóstico: niveles bajos de biotinidasa en suero.
Errores congénitos del metabolismo de las organelas celulares
Enfermedades lisosomales
Mucopolisacaridosis (MPS)
Son un grupo de enfermedades hereditarias de depósito lisosomal caracterizadas por déficit de enzimas involucrados en la degradación de los glicosaminoglicanos. El depósito y acumulación de estos productos en los diferentes órganos y tejidos produce las manifestaciones clínicas de estos pacientes. La herencia es autosómica recesiva, excepto la MPS II Hunter que es recesiva ligada al cromosoma X.
- Clínica sistémica: caras toscas, retraso del crecimiento, deformaciones esqueléticas, compresión medular, problemas cardiacos y respiratorios. En algunos casos hay retraso mental asociado.
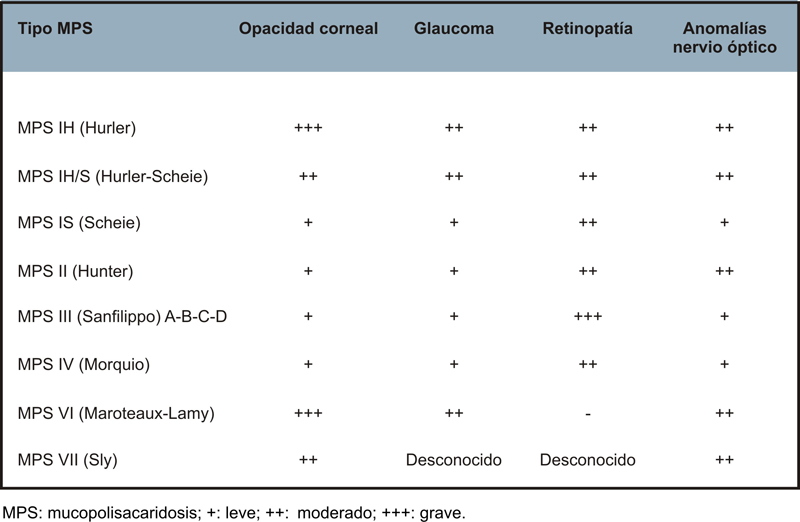
- Manifestaciones oftalmológicas: opacidad corneal, glaucoma, retinopatía y neuropatía óptica. Todos los tipos de MPS pueden presentar edema de papila y secundariamente atrofia óptica. Aproximadamente la mitad de los pacientes con MPS IH, MPS IH/S y MPS VI presentan edema del disco óptico, pero solo entre un 8-19% desarrolla una atrofia óptica. La afectación del nervio óptico es secundaria a diferentes mecanismos: papiledema en los pacientes con aumento de la presión intracraneal, compresión del nervio por engrosamiento de la dura y/o la esclera, y depósito de glicosaminoglicanos en las propias células ganglionares. La atrofia óptica se puede atribuir a la retinopatía por depósito de glicosaminoglicanos en las células del epitelio pigmentario de la retina (EPR) y en la matriz entre los fotorreceptores, lo que conduce a la degeneración retiniana y pérdida de los fotoreceptores. La retinopatía suele evolucionar de forma muy lenta y presentarse de forma tardía, excepto en la MPS III Sanfilippo que la retinopatía es una característica muy constante. En la Tabla 2, se resumen las manifestaciones oftalmológicas típicas de cada tipo de mucopolisacaridosis.
- Diagnóstico: se realiza mediante sospecha clínica, determinación del aumento de glicosaminoglicanos en orina, estudio de la actividad enzimática en plasma o fibroblastos y finalmente por confirmación del defecto genético.
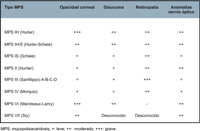
Tabla 2. Manifestaciones oftalmológicas según el tipo de MPS.
Mucolipidosis
Las mucolipidosis son enfermedades de depósito lisosomal de herencia autosómica recesiva. Se han descrito cuatro tipos: I o sialidosis, II, III y IV. La mucolipidosis tipo IV se caracteriza por deterioro neurológico y alteraciones oftalmológicas como opacidad corneal, distrofia retiniana, atrofia óptica y estrabismo.
- Diagnóstico: una vez establecida la sospecha clínica se realiza un estudio enzimático y la confirmación genética.
Esfingolipidosis
Enfermedad de Krabbe
Enfermedad lisosomal que afecta la sustancia blanca del sistema nervioso central y periférico.
- Herencia: autosómica recesiva
- Base molecular: mutación del gen galactosilceramidasa en el cromosoma 14q31.
- Clínica sistémica: la mayoría de los casos debutan a los 6 meses de vida, es la forma infantil o clásica. Cursa con irritabilidad extrema, espasticidad y retraso del desarrollo, con progresivo deterioro motor y mental severo. En ocasiones se acompañan de convulsiones. Entre un 10-15% de los pacientes presenta un inicio tardío de la enfermedad con un curso menos agresivo y de progresión más lenta.
- Clínica oftalmológica: La atrofia óptica es un rasgo muy constante en estos pacientes y es secundaria a desmielinización de la vía óptica, también presentan nistagmo.
- RM cerebral: afectación bilateral de la sustancia blanca, sobre todo en la región parieto-occipital.
- Diagnóstico: determinación de la actividad de la enzima galactosilceramidasa en leucocitos y mediante estudio genético.
Leucodistrofia metacromática
- Herencia: autosómica recesiva.
- Base molecular: mutación del gen de la arilsulfatasa A (ARSA) en el cromosoma 22q13.
- Clínica sistémica: las manifestaciones son heterogéneas y se han descrito 5 formas. La forma infantil tardía se presenta entre el primer y segundo año de vida con problemas en la marcha, alteraciones del habla, espasticidad y deterioro intelectual. También, presentan episodios inexplicables de fiebre y dolor abdominal.
- Clínica oftalmológica: un tercio de los pacientes con la forma infantil tardía tienen atrofia óptica aunque la deficiencia visual cerebral también contribuye a la afectación visual. En ocasiones presentan estrabismo, y menos frecuentemente, depósitos granulares de pigmento en la mácula, alteraciones en la membrana limitante interna, atenuación arteriolar y mácula rojo cereza. El ERG suele ser normal.
- RM cerebral: alteración sustancia blanca, especialmente periventricular.
- Diagnóstico: mediante el estudio de la actividad de arilsulfatasa A en leucocitos y confirmación genética.
Gangliosidosis
Son un conjunto de enfermedades hereditarias de depósito lisosomal de gangliósidos, sobre todo en las neuronas. Existen varias formas, pero se mencionan aquellas que provocan con mayor frecuencia atrofia óptica por el acúmulo de los gangliósidos en las células ganglionares de las retina. Esto produce la típica imagen en mácula rojo cereza y posterior atrofia de estas células. El diagnóstico se realiza una vez establecida la sospecha clínica, con el estudio enzimático y la confirmación genética.
- GM1- gangliosis tipo I o infantil: herencia autosómica recesiva, es producida por la mutación del gen GLB1 en el cromosoma 3p22. Provoca un grado variable de deterioro neurológico y anomalías esqueléticas. Alguna forma presenta visceromegalia. El 50% de los pacientes presentan mancha rojo cereza.
- Enfermedad de Tay-Sachs: herencia autosómica recesiva, es debida a la mutación del gen HEXA en el cromosoma 15q23. Presentan retraso del desarrollo, y progresivamente se instaura la parálisis, demencia y ceguera. Prácticamente la totalidad de los pacientes presentan la mancha rojo cereza.
- Enfermedad de Sandhoff: herencia autosómica recesiva, es producida a la mutación del gen HEXB en el cromosoma 15q13. Es clínicamente indistinguible de la enfermedad de Tay-Sachsy y la totalidad de los pacientes suelen presentar mancha rojo cereza.
Lipofuscinosis neuronal ceroidea
Bajo este término se agrupan unas enfermedades hereditarias de depósito lisosomal que constituyen la principal causa de enfermedad neurodegenerativa en la infancia. Su presentación clínica y las anomalías genéticas son heterogéneas. La forma juvenil (LNCJ) o enfermedad de Batten es la lipofuscinosis neuronal ceroidea más frecuente.
- Herencia: autosómica recesiva en la mayoría de las formas, excepto la que debuta en la edad adulta que es autosómica dominante.
- Base molecular: mutación del gen CLN3 en el cromosoma 16p12.
- Manifestaciones sistémicas: convulsiones, deterioro motor, problemas del habla y demencia. Con el tiempo, pueden aparecer problemas cardiacos.
- Manifestaciones oftalmológicas: la primera alteración en el fondo de ojo es un aspecto granular sutil en el EPR de la mácula, posteriormente, se observa atrofia óptica, atenuación del árbol vascular y acúmulos de pigmento en la retina periférica. El ERG está muy alterado o abolido, aunque inicialmente se altera la onda “b”, pero no la onda “a”. La pérdida visual suele ser el síntoma de presentación en la forma juvenil hacia los 6 ó 7 años. Las formas infantil e infantil tardía presentan una clínica neurológica muy florida y casi nunca la afectación visual es la primera manifestación.
- RM cerebral: normal en niños menores de 10 años.
- Diagnóstico: visualización de los depósitos de lipofuscina en la biopsia cutánea y mediante confirmación genética.
Enfermedades peroxisomales
Las enfermedades que afectan a los peroxisomas se han dividido en tres grupos:
Enfermedades secundarias a la alteración en la biogénesis de los peroxisomas
Síndrome de Zellweger, adrenoleucodistrofia neonatal y enfermedad infantil de Refsum. Todas ellas son de herencia autosómica recesiva. Las manifestaciones sistémicas del síndrome de Zellweger son hipotonía severa, convulsiones y dismorfismo cráneofacial. La adrenoleucodistrofia neonatal se caracteriza por hipotonía y convulsiones sin características dismórficas, presentan atrofia cortical adrenal pero raramente con insuficiencia adrenal. La enfermedad de Refsum infantil es la forma menos severa de este grupo de enfermedades peroxisomales. Las principales manifestaciones oftalmológicas son la retinopatía pigmentaria con ERG abolido y la atrofia óptica secundaria a la retinopatía pero también a la desmielinización del nervio óptico. También pueden presentar opacidad corneal, cataratas y glaucoma congénito, pero con menor frecuencia y sobre todo en pacientes con el síndrome de Zellweger.
Enfermedades por déficit de múltiples enzimas
Condrodisplasia punctata, tipo rizomélico. Su herencia es autosómica recesiva. Sistémicamente presentan extremidades proximales cortas, dermatitis y retraso psicomotor. Su principal manifestación oftalmológica es la catarata capsular anterior bilateral.
Enfermedades producidas por déficit de un único enzima
Adrenoleucodistrofia ligada al cromosoma X, hiperoxaluria tipo 1 primaria y enfermedad de Refsum clásica.
- La adrenoleucodistrofia ligada al cromosoma X, en su forma infantil, suele presentarse durante la primera década de la vida con labilidad emocional e hiperactividad. Posteriormente, se añaden más alteraciones neurológicas con regresión psicomotora, la hipofunción adrenal aparecen en todos los casos. La clínica oftalmológica se caracteriza por pérdida de visión secundaria a la desmielinización de la vía óptica y es una característica muy constante. El ERG es normal.
- La hiperoxaluria tipo 1 primaria es autosómica recesiva. Suele manifestarse en la infancia con fallo renal y nefrolitiasis. También pueden presentar osteodistrofia que provoca fracturas patológicas con traumatismos mínimos. La clínica oftalmológica consiste en alteración del EPR por depósito de cristales de oxalato cálcico en el polo posterior de forma bilateral y simétrica, pero sin alteración visual. Se ha descrito atrofia óptica en algunos pacientes, posiblemente secundaria al aumento de la presión intracraneal por la presencia de cristales en el líquido cefalorraquídeo.
- La enfermedad de Refsum tipo clásica es autosómica recesiva y suele debutar entre la primera y tercera década de la vida. Sistémicamente se manifiesta con sordera, neuropatía periférica y ataxia, además, pueden presentar cardiopatía, malformaciones primarias de los metatarsos y metacarpos y cambios cutáneos similares a la ictiosis. La manifestación oftalmológica consiste en ceguera nocturna secundaria a retinosis pigmentaria, muchas veces es la primera manifestación de la enfermedad.
Enfermedades mitocondriales
En este apartado solo se incluyen las enfermedades mitocondriales sindrómicas, es decir, aquellas con síntomas neurológicos o sistémicos y con atrofia óptica como una de sus manifestaciones.
Síndrome de Leigh
- Herencia: autosómica recesiva y mitocondrial.
- Base molecular: es de una gran heterogeneidad genética ya que se han detectado mutaciones tanto en genes nucleares como mitocondriales que participan en el metabolismo oxidativo.
- Clínica sistémica: se trata de una enfermedad neurodegenerativa progresiva de inicio precoz. La clínica neurológica consiste en hipotonía, espasticidad, corea y otros trastornos del movimiento, ataxia cerebelosa, neuropatía periférica y problemas respiratorios por la disfunción del tronco del encéfalo. El hallazgo bioquímico más constante es la elevación de lactato y piruvato en sangre.
- Clínica oftalmológica: atrofia óptica, retinopatía pigmentaria, nistagmo y oftalmoplejía extrínseca crónica progresiva.
- RM cerebral: lesiones localizadas en ganglios de la base y el tronco encéfalo.
Ataxia Friedreich
Es la causa más frecuente de ataxia hereditaria recesiva. Es una enfermedad neurodegenerativa que cursa con ataxia progresiva, disartria, pérdida de reflejos tendinosos profundos y de la sensibilidad, deformidades en los pies, escoliosis y cardiomiopatía. La neuropatía óptica es muy frecuente aunque los pacientes no refieren pérdida de visión.
Enfermedad de Charcot-Marie-Tooth (CMT)
Es una de las neuropatías periféricas hereditarias más frecuente. Un subtipo de CMT, la neuropatía sensorial y motora tipo VI, se caracteriza por neuropatía periférica axonal y atrofia óptica. La herencia puede ser autosómica recesiva o dominante. La neuropatía periférica precede en una década o más la aparición de la atrofia óptica que se desarrolla en la adolescencia tardía. La clínica oftalmológica consiste en una disminución de la visión subaguda pero importante (<20/400), alteración de la visión de los colores y defectos centrales en el campo visual. Al igual que pasa con algunos casos de neuropatía óptica hereditaria de Leber, algunos pacientes recuperan visión años después del inicio de la neuropatía óptica. Algunos pacientes presentan mutación en el gen de la mitofusina.
Otros síndromes mitocondriales con neuropatía óptica
Síndrome MERRF (epilepsia mioclónica con fibras rojo-rasgadas), síndrome MELAS (encefalopatía mitocondrial, acidosis láctica y episodios similares a accidentes vasculares), CPEO (oftalmoplejía externa progresiva crónica), síndrome de Kearns-Sayre y síndrome MNGIE (neuropatía gastrointestinal con encefalopatía).
Enfermedades neurodegenerativas genéticas sin defecto metabólico conocido
Enfermedad de Canavan
- Herencia: autosómica recesiva. Ocurre principalmente en judíos Ashkenazi.
- Base molecular: mutación del gen de la aspartoacilasa en el cromosoma 17p13.
- Clínica sistémica: Durante los primeros meses de vida, los niños que nacen sanos, presentan un progresivo deterioro neurológico debido a la desmielinización y leucodistrofia cerebral. Aparecen macrocefalia sin hidrocefalia, falta de control cefálico, convulsiones, hipotonía inicial que evoluciona a espasticidad tardía, retraso importante del desarrollo y retraso mental grave.
- Clínica oftalmológica: atrofia óptica.
- RM cerebral: afectación bilateral difusa de la sustancia blanca, y en estadios tardíos, atrofia cortical.
- Diagnóstico: detectar disminución de la actividad del aspartoacilasa en fibroblastos de la piel o aumento de ácido N-acetilaspártico en orina.
Enfermedad de Pelizaeus-Merzbacher
- Herencia: recesiva ligada al cromosoma X.
- Base molecular: mutación del gen que codifica la proteína 1 del proteolípido.
- Clínica sistémica: es una leucodistrofia caracterizada por retraso psicomotor, hipotonía, espasticidad y retraso mental variable. Se clasifica en tres formas según la edad de aparición y la gravedad del cuadro: connatal, transitoria y clásica. Durante los primeros meses de vida presentan nistagmo rotatorio y movimientos de la cabeza que permite diferenciarlas de otras leucodistrofias, aunque esta manifestación desaparecerá con el tiempo.
- Clínica oftalmológica: nistagmo en fase inicial y atrofia óptica que suele aparecer de forma más tardía.
- RM cerebral: hipomielinización de grado variable según la forma clínica.
- Diagnóstico: estudio genético.
Síndrome de PEHO
Encefalopatía progresiva con edema, hipsarritmia y atrofia óptica.
- Herencia: autosómica recesiva. Suelen tener ascendencia finlandesa.
- Base molecular: no se ha determinado el defecto metabólico causante de este síndrome.
- Clínica sistémica: los niños nacen sanos o con ligera hipotonía, entre las dos semanas y los tres meses de vida aparece hipotonía progresiva, baja visión y sacudidas en las extremidades. Otras manifestaciones son los espasmos infantiles, reflejos tendinosos profundos exagerados y se detiene el desarrollo psicomotor. Es muy característico el edema subcutáneo sin fóvea en la cara y extremidades. Los dedos de las manos son cónicos y la cara suele ser dismórfica con epicantus, hipoplasia tercio medio facial, lóbulos de las orejas prominentes, hipertrofia gingival y mentón pequeño.
- Clínica oftalmológica: atrofia óptica de aparición precoz, y a los dos años de edad, ceguera y nistagmo.
- RM cerebral: hipoplasia del cerebelo. Con el tiempo aparece atrofia del tronco, cerebelo y nervios ópticos. Puede también observarse mielinización anormal sugestiva de leucomalacia periventricular.
- Diagnóstico: estudio genético.
Disautonomía familiar
- Herencia: autosómica recesiva. Suele afectar a judíos Ashkenazi.
- Base molecular: mutación del gen IKBKAP en el cromosoma 9q31.
- Clínica sistémica: presentan sobre todo disfunción autonómica, pero también se afectan nervios motores y sensitivos periféricos. Se presenta con pobre reflejo de succión, hipotonía, hipotermia y regurgitación. También, se destaca la ausencia de las papilas fungiformes de la lengua y disminución muy marcada del sentido del gusto.
- Manifestaciones oftalmológicas: hipoestesia corneal y alacrimia. Otras manifestaciones oftalmológicas son tortuosidad vascular, ptosis, anisocoria, exotropía y miopía. La atrofia óptica se ha descrito muy infrecuentemente.
Degeneraciones espinocerebelosas
Se han identificado al menos 30 subtipos con mutaciones en diferentes genes, la mayoría con herencia autosómica dominante y debut en la edad adulta. Se caracterizan por ataxia junto con otros signos neurológicos, incluyendo trastornos oculomotores, defectos cognitivos, disfunción piramidal y extrapiramidal, y la afectación del sistema nervioso bulbar, espinal y periférico. Algunos subtipos cursan con retinopatía, como la ataxia espinocerebelosa tipo 7 (SCA 7), mientras que la SCA 1 presenta atrofia óptica.
La neurodegeneración con acumulo cerebral de hierro (NBIA)
Abarca un grupo de trastornos neurodegenerativos que se caracterizan por un depósito de hierro en los ganglios de la base y otras regiones del cerebro.
NBIA1
Se puede clasificar en tres categorías: enfermedad clásica (inicio precoz y curso rápidamente progresivo), enfermedad atípica (inicio más tardío y curso de progresión más lento), y enfermedad intermedia (con características de las dos anteriores, por ejemplo, inicio tardío con un curso rápidamente progresivo)
- Herencia: autosómica recesiva.
- Base molecular: mutación del gen pantotenakinasa (PANK2), Se localiza en el cromosoma 20p13, mutada en todos los casos de la forma clásica y en un tercio de la forma atípica.
- Clínica sistémica: distonía, parkinsonismo, coreoatetosis, afectación la vía cortico-espinal y deterioro cognitivo. La distonía sobre todo se aprecia en las formas de inicio precoz y el parkinsonismo en las formas de inicio en la edad adulta. Las alteraciones del lenguaje y los trastornos psiquiátricos aparecen en las de inicio tardío.
- Manifestaciones oftalmológicas: atrofia óptica y retinopatía pigmentaria, esta última es característica de la forma clásica.
- RM cerebral: todos los pacientes muestran el signo del “ojo de tigre” en los ganglios de la base, aunque no es patognomónico.
NBIA2A y NBIA2B
- Herencia: autosómico recesiva.
- Base molecular: mutación del gen PLA2G6 en el cromosoma 22q13.
- Clínica sistémica: suele debutar a los dos años de edad con deterioro mental y motor, signos de afectación la vía piramidal bilateral y marcada hipotonía.
- Manifestaciones oftalmológicas: el deterioro visual es precoz, con atrofia óptica en un alto porcentaje de los casos a los tres años.
Síndrome GAPO
- Herencia: autosómico recesiva.
- Base molecular: mutación del gen ANTXR1 en el cromosoma 2p13.
- Clínica sistémica: retraso crecimiento, alopecia y pseudoanodoncia (ausencia de dientes por retención o erupción tardía).
- Manifestaciones oftalmológicas: atrofia óptica progresiva, aunque no todos los pacientes la presentan.
Diagnóstico
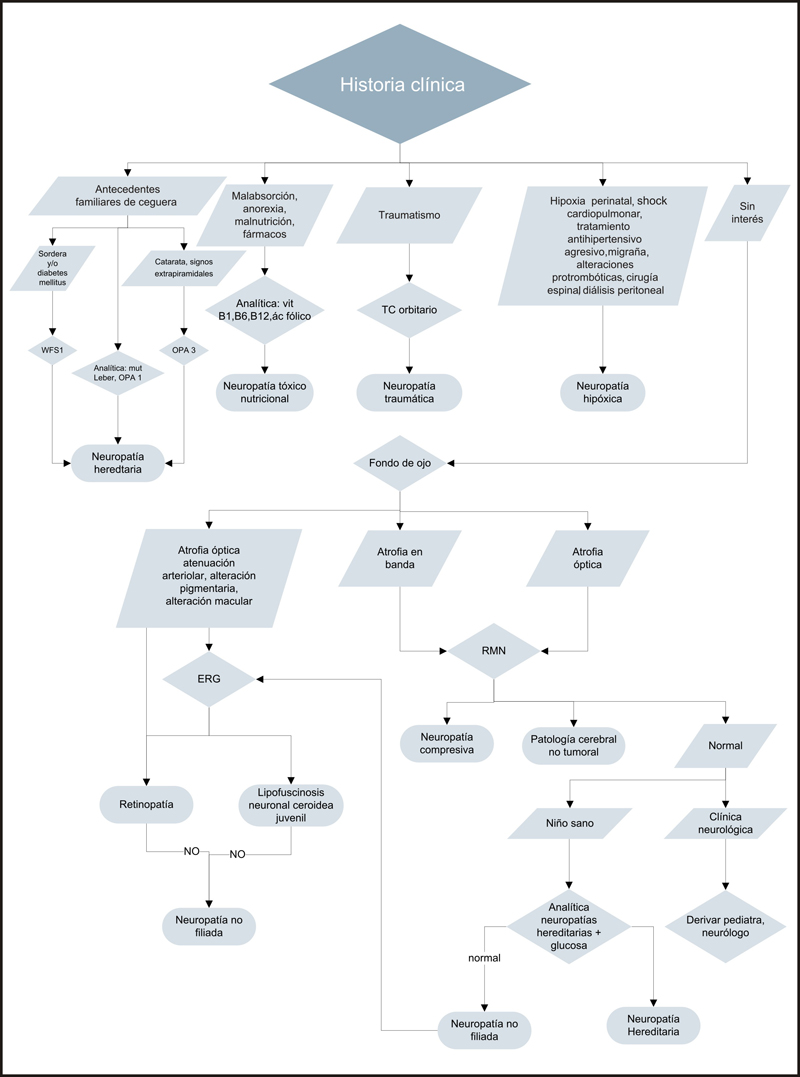
Ante una atrofia óptica bilateral en el niño es imprescindible una historia clínica detallada para intentar orientar el cuadro y solicitar las pruebas complementarias adecuadas.
- Edad de aparición de la pérdida de visión, muy difícil de establecer en niños pequeños.
- Curso de la clínica: estable o progresiva.
- Antecedentes gestacionales como ingesta de alcohol o drogas , y exposición a tóxicos
- Consanguinidad de los progenitores
- Antecedente de prematuridad, hipoxia-isquemia perinatal, patología intracraneal (hemorragia intraventricular, meningoencefalitis, etcétera).
- Antecedente de patología digestiva con malabsorción, diabetes mellitus y/o sordera.
- Traumatismos cráneo-faciales recientes.
- Tratamientos actuales o recientes.
- Antecedentes familiares de ceguera.
La exploración oftalmológica tendría que ser lo más completa posible de acuerdo con el grado de colaboración del paciente:
- Agudeza visual.
- Visión de colores.
- Motilidad ocular extrínseca e intrínseca.
- Presencia o no de nistagmo.
- Fondo de ojo bajo midriasis. El aspecto del disco de óptico proporciona mucha información, dado que si la palidez es segmentaria podría orientar sobre la etiología de la atrofia. La atrofia en banda hace sospechar patología compresiva quiasmática. La palidez temporal es característica de la atrofia óptica dominante y la neuropatía tóxico-nutricional. Por otro lado, se debe explorar bien la periferia y aspecto de los vasos retinianos que hace sospechar que la atrofia óptica es secundaria a una retinopatía.
- El campo visual se suele poder realizar en niños mayores de 8 años, siempre que no presenten patología neurológica asociada.
- Tomografía de coherencia óptica, podría ser útil, para valorar que sectores papilares están más afectos y ayudar en el diagnóstico.
- ERG fotópico y escotópico son imprescindibles para establecer el diagnostico cuando se sospecha una retinopatía subyacente.
Las pruebas complementarias sistémicas se deberían solicitar en función de la historia y exploración oftalmológica: RM cerebral, estudio metabólico, determinación de vitaminas (B1, B6 y B12) y ácido fólico, estudio de las mutaciones en DNA mitocondrial, gen OPA1 y WFS1, glicemia y audiometría. Los pacientes con clínica neurológica deberán ser valorados por un neurólogo pediátrico.
El protocolo diagnóstico se resume en forma de algoritmo (Algoritmo 1).
Bibliografía
- Hereditary Optic Neuropathies. En: Walsh & Hoyt’s. Clinical Neuro-Ophtalmology 6th ed. Philadelphia: Lippincott Williams & Wilkins, 2005;465-501.
- Van Effenterre R, Boch AL. Craniopharyngioma in adults and children: a study of 122 surgical cases. J Neurosurg. 2002;97:3-11.
- Warmuth-Metz M, Gnekow AK, Müller H, Solymosi L. Differential diagnosis of suprasellar tumors in children. Klin Padiatr. 2004:216:323-30.
- Barboni P, Savini G, et al. Leber's hereditary optic neuropathy with childhood onset. Invest Ophthalmol Vis Sci. 2006;47:5303-9.
- Optic Atrophy in children. En: Browsky MC, ed. Pediatric neuro-ophthalmology. Rochester: Springer, 2010;155-2011
- Imperia PS, Lazarus HM, Lass JH. Ocular complications of systemic cancer chemotherapy. Surv Ophthalmol. 1989;34:209-30.
- Nambiar S, Rellosa N, et al. Linezolid-associated peripheral and optic neuropathy in children. Pediatrics. 2011;127:e1528-e1532.
- Sánchez-Dalmau B, Vela MD. Neuropatías ópticas nutricionales y tóxicas. En: Arruga Ginebreda J, Sánchez Dalmau B, ed. Neuropatías ópticas: diagnóstico y tratamiento. LXXVIII Ponencia oficial de la sociedad española de oftalmología 2002.
- Ashworth JL, Kruse FE, et al. Ocular manifestations in the mucopolysaccharidoses – a reiew. Clin Experiment Ophthalmol. 2010;38:12-22.
- Ashworth JL, Biswas S, Wraith E, Lloyd IC. Mucopolysaccharidoses and the eye. Surv Ophthalmol. 2006;51:1-17.
- Folz SJ, Trobe JD. The peroxisome and the eye. Surv Ophthalmol. 1991;35:353-68.
- Fraser JA, Biousse V, Newman NJ. The neuro-ophthalmology of mitochondrial diseases. Surv Ophthalmol. 2010;55:299-334.
- DeBrosse S, Parikh S. Neurologic disorders due to mitochondrial DNA mutations. Semin Pediatr Neurol. 2012;19:194-202.
- Stricker S, Oberwahrenbrock T, et al. Spinocerebellar ataxia type 1. PLos ONE. 2011; 6: e23024.
- Bozorg S, Ramirez-Montealegre D, Chung M, Pearce DA. Juvenile neuronal ceroid lipofuscinosis (JNCL) and the eye. Surv Ophthalmol. 2009;54:463-71.
- Williams ZR, Hurley PE, et al. Late onset optic neuropathy in methylmalonic and propionic academia. Am J Ophthalmol. 2009;147:929-33.
- Tsina EK, Marsden DL, Hansen RM, Fulton AB. Maculopathy and retinal degeneration in cobalamin C methylmalonic aciduria and homocystinuria. Arch Ophthalmol. 2005;123:1143-6.
- Ford RL, Lee V, Xing W, Bunce C. A 2-year prospective surveillance of pediatric traumatic optic neuropathy in the United Kingdom. J AAPOS. 2012;16:413-7.
- Goldenberg-Cohen N, Miller NR, Repka MX. Traumatic optic neuropathy in children and adolescents. J AAPOS. 2004; :20-7.
-
Yu-Wai-Man P, Griffiths PG. Steroids for traumatic optic neuropathy. The Cochrane Library. 2011, Issue 1.