2Hospital Sant Joan de Déu. Esplugues de Llobregat. Barcelona.
CORRESPONDENCIA
RESUMEN
RESUM
ABSTRACT
Introducción
La osteogénesis imperfecta (OI) es una patología genética hereditaria del tejido conectivo, resultante de mutaciones en los genes que codifican el colágeno tipo I. Se caracteriza por fragilidad ósea y susceptibilidad a fracturas, cuya severidad es variable: desde el sutil aumento en la frecuencia de las fracturas, a las fracturas prenatales1. La primera descripción científica de OI fue realizada en 17882. Se manifiesta en los tejidos en los que la proteína de la matriz principal es colágeno de tipo I, es decir, en el hueso, los ligamentos, los dientes, y la esclerótica. Las manifestaciones clínicas clásicas incluyen múltiples fracturas óseas que conducen a baja estatura, sordera, escleras azules, y articulaciones flexibles (hipermovilidad, pie plano y mala dentición). Por otra parte, las formas más severas de OI pueden ocasionar piernas y brazos arqueados, cifosis y escoliosis.Epidemiologia
La OI tiene una prevalencia de aproximadamente 6-7/100.000 nacimientos. La prevalencia e incidencia varía según el tipo de osteogénesis, el tipo I y el tipo IV representan más de la mitad de todos los casos3.Patogénesis
La OI comprende un grupo heterogéneo de trastornos. Se estima que 90% de los casos, es producido por una variación de los genes COL1A1 o COL1A2 y el 10% restantes por una variación recesiva en 8 genes desconocidos por el momento. Además, se ha identificado una deleción de 0.5 kb en un alelo de la cadena pro alfa 1, que produce una desintegración en la biosíntesis de colágeno tipo I. Los mecanismos patogénicos restantes todavía no están completamente dilucidados4.
La osteogénesis imperfecta se puede clasificar en 4 tipos (I-IV de Sillence) según la herencia5:- Tipo I (herencia autosómica dominante): leve. Escleras azules.
- Tipo II (herencia autosómica recesiva): letal. Perinatal, se clasifica en A, B y C dependiendo de los hallazgos radiológicos.
- Tipo III (herencia autosómica recesiva): progresivo. Deformante.
- Tipo IV (herencia autosómica dominante): esclera normal.
Clínica oftalmológica
La manifestación ocular más frecuente consiste en la presencia de escleróticas azules. Estas, se asocian con la reducción de rigidez escleral (adelgazamiento de las fibras que componen la esclerótica lo que permite la transparencia de los vasos coroideos) (Figura 1).

Figura 1. Escleras azules en un paciente con osteogénesis imperfecta.
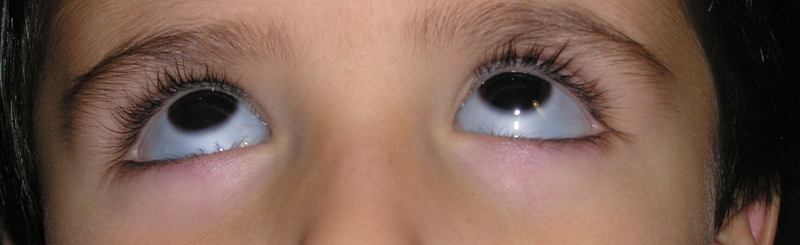
Este signo es más evidente con luz natural.
Los pacientes con OI pueden desarrollar miopía alta por la elongación axial del globo ocular y el desarrollo de estafiloma posterior3. Además, aparecen membranas neovasculares atribuibles a roturas espontáneas en la membrana del epitelio pigmentario de la retina, membrana de Bruch y complejo coriocapilar epitelio-pigmentario6. Lo anterior puede conducir a un aumento de la susceptibilidad al desprendimiento de retina y a hemorragias retinianas7.
Ahora bien, las fibras de las capas de colágeno interna y externa de la membrana de Bruch contienen los tipos I, III y V de colágeno, y las de la membrana basal coriocapilar contienen colágeno tipo IV8, por lo tanto, la aparición de membranas neovasculares también se puede atribuir a la anormalidad primaria de sus capas de colágeno en la OI, lo cual afecta su integridad estructural. Por otra parte, la fragilidad de la membrana de Bruch debido la mineralización de sus fibras elásticas predispone al desarrollo grietas (estrías angiodes). El juego de las fuerzas ejercidas por los músculos extraoculares en el ojo y el efecto de la inmovilización del nervio óptico, modula la orientación centrífuga de las estrías radialmente alrededor del disco del nervio óptico9 (Figura 2). Hallazgos oculares adicionales incluyen glaucoma, queratocono, opacidad de la córnea, microcórnea y agenesia congénita de la capa de Bowman10-12.
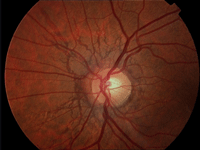
Figura 2. Estrías angioides en el fondo de ojo de un paciente con osteogénesis imperfecta.
Diagnóstico13,14
El diagnóstico prenatal se realiza por ultrasonografía en los tipos II y III, apreciándose fracturas prenatales y aumento de la translucencia. Las fracturas pueden detectarse desde las 14 semanas de embarazo en el tipo II y desde la semana 18 en el tipo III. (Figuras 3 y Figura 4). Las múltiples fracturas producen deformidad de huesos largos, costillas y cráneo. En el tipo IV ocasionalmente se realiza el diagnóstico sobre la semana 22 por acortamiento de huesos largos, sin fracturas ni osteopenia. La OI tipo I no se puede diagnosticar in utero.
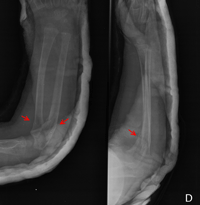
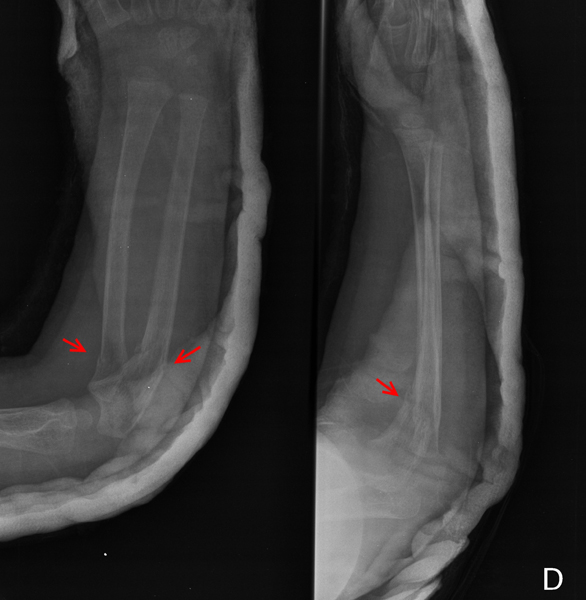
Figura 3. Fractura de radio y cúbito (flechas).
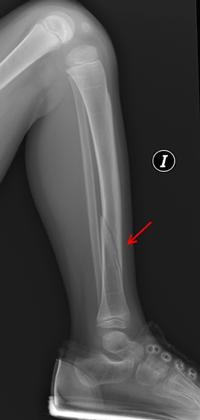
Figura 4. Fractura distal de tibia (flecha) en el mismo paciente afecto
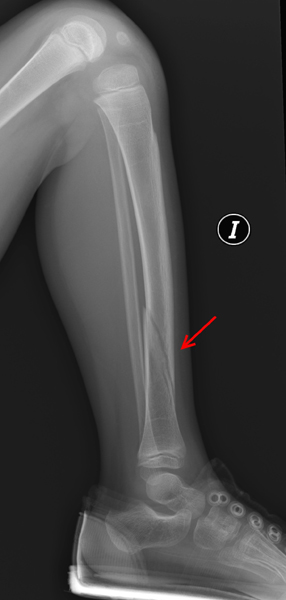
de osteogenesis imperfecta tipo 1.
Los hallazgos ecográficos pueden ser confirmados en el laboratorio mediante biopsia de vellosidad coriónica con cultivo o a través de paracentesis de vellosidad coriónica, determinándose la producción anómala de colágeno. Además, se puede realizar estudio molecular y genético de ADN. La confirmación de laboratorio de OI en este caso se realiza preferiblemente mediante el análisis de ADN de los genes implicados en OI, o por la disminución o anormal producción de (pro) colágeno de tipo I por los fibroblastos, medidos mediante electroforesis.
El diagnóstico post natal se puede realizar en todos los tipos de OI mediante una historia familiar positiva, presencia de escleras azules, osteoporosis, múltiples deformidades en el cráneo y platispondilia15. El diagnóstico diferencial se establece con la displasia campomélica y la hipofosfatasia letal perinatal por los hallazgos similares.
Tratamiento
Cuando se ha establecido el diagnóstico de OI, el paciente preferentemente debe ser evaluado por un equipo multidisciplinario3. Los miembros del equipo serían (como mínimo) cirujanos ortopédicos, endocrinólogos, fisioterapeutas, pediatras y especialistas en rehabilitación. Se debe solicitar valoración por otras disciplinas según las necesidades del paciente.El tratamiento consiste en manejo farmacológico, ortopédico, dental, medicina física, manejo de la pérdida de audición; y la prevención primaria (fondo de ojo seriado para descartar papiledema) y secundaria (desprendimiento de retina, membrana neovascular subretiniana)5.
Con relación al manejo farmacológico, se prescriben los bifosfonatos orales e intravenosos para todos los tipos de OI, tanto en niños como en adultos16. El uso de las hormonas de crecimiento para tratar la baja estatura en los tipos III y IV de OI todavía está bajo investigación activa17.
Tratamiento ortopédico
En caso de disminución de la mineralización ósea, fracturas de alta frecuencia y /o deformidades de los huesos, se pueden utilizar varillas intramedulares en la mayoría de los pacientes con OI tipo III y IV, y algunas veces en la OI tipo I18.Tratamiento de medicina física (rehabilitación)
Es necesario un programa de rehabilitación intensiva sobre todo en los tipos de OI III y IV18, con principios de intervención tales como la colocación correcta del niño, apoyo adecuado, fortalecimiento muscular (isotónico) y el acondicionamiento aeróbico19.Tratamiento dental
En los pacientes con OI, las fracturas y el desgaste excesivo de los dientes se produce a menudo. Puede ser tratado con el recubrimiento de estos mediante polímeros duros con el fin de prevenir las infecciones y deformidades faciales debido a la pérdida de los mismos y/o maloclusión18.Tratamiento para la pérdida de audición
La pérdida de audición es común en los pacientes con OI. Inicialmente es una pérdida conductiva, sin embargo el deterioro auditivo progresa, y finalmente emerge un importante componente neurosensorial. Se recomienda la valoración después de la adolescencia cada 3-5 años5. Inicialmente, los audífonos serán suficientes. A medida que la pérdida de la audición progresa, la estapedectomía puede ser considerada, sin embargo, a largo plazo la restauración de la audición puede ser insatisfactoria debido a la fragilidad de las estructuras del oído medio osicular. El implante coclear es buena opción ante la pérdida neurosensorial, pero los datos son demasiado limitados para extraer conclusiones sobre su eficacia19.
Invaginación basilar
Se trata de una complicación rara que ocurre en adultos con OI tipo III cuando la parte superior de la vértebra C2 migra ascendentemente hacia el cráneo lo cual puede conducir al cierre del foramen magnum ocasionando hidrocefalia, presión sobre el tronco cerebral y hernia posterior del cerebro requiriendo colocación de una derivación ventricular o cirugía. Sólo la inmovilización prolongada mediante ortesis ha demostrado estabilizar los síntomas y la progresión18.Control oftalmológico
Los pacientes con OI Tipo I que son el grupo con mayor compromiso ocular deben ser valorados anualmente para seguimiento y prevención secundaria.Conclusión
La osteogénesis imperfecta es una enfermedad hereditaria compleja con una notable variabilidad clínica que debemos enmarcar en las diferentes formas de presentación. Se precisa más investigación en la detección de genes que codifican las proteínas implicadas en la biosíntesis de colágeno de tipo I. Estos pacientes requieren un manejo multidisciplinario.Ante un paciente con escleras azules se debe investigan los antecedentes familiares y descartar la presencia de fracturas óseas de repetición que puedan sugerir la presencia de esta enfermedad.
Bibliografía
- Kocher M, Shapiro F. Osteogenesis imperfecta. J Am Acad Orthop Surg. 1998;6:225-36.
- Pelthier L. The classic: congenital osteomalacia. Olaus Jacob Ekman. Clin Orthop Relat Res. 1981;159:3-5.
- Steiner R, Adsit J, Basel D. COL1A1/2-Related Osteogenesis Imperfecta. Pagon RA, Adam MP, Bird TD, et al., Ed. GeneReviewsTM [Internet]. Seattle Univ. Washington, Seattle; 1993-2014. Disponible en: http://www.ncbi.nlm.nih.gov/books/NBK1295/
- Sillence D, Senn A, Danks D. Genetic heterogeneity in osteogenesis imperfecta. J Med Genet. 1979;16:101-16.
- Chu M, et al. Internal deletion in a collagen gene in a perinatal lethal form of osteogenesis imperfecta. Nature. 1983;304:79-80.
- Scott A, Kashani S, Towler H. Progressive myopia due to posterior staphyloma in type 1 osteogenesis imperfecta. Int Ophthalmol. 2005;26:167-9.
- Ganesh A, Jenny C, Geyer J, Shouldice M, Levin A. Retinal hemorrhages in type 1 osteogenesis imperfecta after minor trauma. Ophthalmology. 2004;111:1428-31.
- Marshall G, Konstas A, Lee W. Collagens in ocular tissues. Br J Ophthalmol. 1993;77:515-24.
- Rishi P, Rishi E, Venkatraman A. Intravitreal bevacizumab for treatment of choroidal neovascularization associated with osteogenesis imperfecta. Indian J Ophthalmol. 2012;60:229-31.
- Evereklioglu C, et al. Central corneal thickness is lower in osteogenesis imperfecta and negatively correlates with the presence of blue sclera. Ophthalmic Physiol Opt. 2002;22:511-5.
- Gorovoy MS, Gorovoy IR, Ullman S, Gorovoy JB. Descemet stripping automated endothelial keratoplasty for spontaneous descemet membrane detachment in a patient with osteogenesis imperfecta. Cornea. 2012;31:832-5.
- Rosbach J, Vossmerbaeumer U, Renieri G, Pfeiffer N, Thieme H. [Osteogenesis imperfecta and glaucoma. A case report]. Ophthalmologe. 2012;109:479-82.
- Viora E, et al. Increased nuchal translucency in the first trimester as a sign of osteogenesis imperfecta. Am J Med Genet. 2002;109:336-7.
- Marini J, et al. Consortium for osteogenesis imperfect mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat. 2007b;28:209-221.
- Chapman S, Hall C. Non-accidental injury or brittle bones. Pediatr Radiol. 1997;27.
- Glorieux F, et al. Cyclic administration of pamidronate in children with severe osteogenesis imperfecta. N Engl J Med. 1998;339:947-52.
- Marini J, et al. Positive linear growth and bone responses to growth hormone treatment in children with types III and IV osteogénesis imperfecta: high predictive value of the carboxyterminal propeptide of type I procollagen. J Bone Min Res. 2003;18:237-43.
- Monti E, et al. Current and emerging treatments for the management of osteogenesis imperfecta. Ther Clin Risk Manag. 2010;6:367-81.
- Marini J, Cabral W, Barnes A. Null mutations in LEPRE1 and CRTAP cause severe recessive osteogenesis imperfecta. Cell Tissue Res. 2010;339:59-70.