Volumen 21 - Número 5 - Diciembre 2013
Retinoblastoma y otros tumores infantiles: diagnóstico y clasificación
J. Català -Mora1, J. DÃaz2, N. MartÃn-Begué3, M. Quintana-Casany4, M. Suñol5
1Responsable de la Unidad de Retinoblastoma. Unidad de Vítreo-Retina. Hospital Sant Joan de Déu. Esplugues de Llobregat. Barcelona. 2Unidad Vítreo-Retina. Hospital Sant Joan de Déu. Esplugues de Llobregat. Barcelona. Hospital Sant Pau. Barcelona
3Unidad Oftalmología Pediátrica. Hospital Universitari Vall d’Hebron, Barcelona. 4Exjefe de servicio de Oftalmología. Hospital Universitario Bellvitge. Barcelona.5Médico Adjunto Servicio Anatomía Patológica. Hospital Sant Joan de Déu. Esplugues de Llobregat. Barcelona.
3Unidad Oftalmología Pediátrica. Hospital Universitari Vall d’Hebron, Barcelona. 4Exjefe de servicio de Oftalmología. Hospital Universitario Bellvitge. Barcelona.5Médico Adjunto Servicio Anatomía Patológica. Hospital Sant Joan de Déu. Esplugues de Llobregat. Barcelona.
CORRESPONDENCIA
Jaume Català-Mora
E-mail: jcatalam@hsjdbcn.org
E-mail: jcatalam@hsjdbcn.org
El retinoblastoma es un tumor maligno que se origina en las células primitivas de la retina sensorial. Es el tumor intraocular más frecuente en la infancia, con una incidencia de 1 por cada 14.000 a 23.000 recién nacidos vivos. El retinoblastoma tiene un fuerte componente genético y se produce por la inactivación de los dos alelos del gen RB1, localizado en la región q14 del cromosoma 13. El RB1 es un antioncogén y la pérdida de su función da lugar a una proliferación celular descontrolada. Un tercio de los pacientes desarrollan enfermedad bilateral, y en ellos existe una predisposición genética a desarrollar diferentes tumores. La edad media en el momento del diagnóstico en los casos bilaterales es de 12 meses, y en los unilaterales de 23 meses. No se han descrito diferencias raciales ni por sexo.
La mayoría de los casos son detectados por el entorno familiar. Sólo un 5% a 7% se detectan en una exploración sistemática por un pediatra. La aparición de cualquiera de estos signos requiere la exploración del niño por un oftalmólogo, con examen de fondo de ojo.
En el examen histológico se aprecia que el retinoblastoma está constituido por células pequeñas, indiferenciadas, de núcleo basófilo e hipercromático, y escaso citoplasma (Figura 3). Se disponen difusamente y se agrupan formando rosetas de Flexner-Wintersteiner (Figura 4) constituidas por células cuboideas, de núcleo basal con proyecciones citoplasmáticas, dispuestas alrededor de una luz central, cuya identificación indica diferenciación. También puede verse alguna área con rosetas más diferenciadas, citológicamente benignas, con abundante citoplasma y núcleo pequeño, denominadas fleurettes (Figura 5).Tanto en los retinoblastomas diferenciados como en los indiferenciados pueden identificarse también rosetas de Homer-Wrigth; aunque éstas no son específicas del retinoblastoma, son semejantes a las rosetas de Flexner-Wintersteiner pero sin luz central.
El retinoblastoma suele mostrar extensas zonas de necrosis, con presencia de calcificaciones; las zonas más preservadas son las que se encuentran alrededor de vasos sanguíneos (Figura 6). También suelen identificarse abundantes figuras de mitosis. Inmunohistoquímicamente las células tumorales expresan marcadores neurales, como enolasa neuronal específica, sinaptofisina y también proteína S100.
La evaluación de la extensión del tumor es indispensable para definir los criterios histológicos de riesgo del retinoblastoma. Deben valorarse la invasión del nervio óptico, el margen de resección y la invasión poslaminar, intralaminar o prelaminar; también la invasión de coroides, de esclerótica y de la órbita, así como la extensión a la cámara anterior.
La evaluación de los criterios histológicos de riesgo ha variado considerablemente a lo largo del tiempo. En la actualidad (Tabla 1) se consideran criterios histológicos de bajo riesgo la invasión de la coroides y de la cámara anterior. Los criterios de riesgo intermedio son la invasión de la esclerótica intraescleral o la invasión del nervio óptico posterior a la lámina cribosa (Figura 7). Como criterios histológicos de alto riesgo se consideran la invasión de la esclerótica transescleral, la invasión del margen quirúrgico del nervio óptico (Figura 8) y la invasión extraocular.
Por último, es muy importante realizar un informe de anatomía patológica estandarizado que incluya todos los hallazgos histológicos que permitan una correcta estadificación del tumor. Así, deben constar el diagnóstico, el patrón de crecimiento, la diferenciación, la invasión de estructuras y otros hallazgos histopatológicos que puedan encontrarse.
La exploración del fondo de ojo debe realizarse con las pupilas dilatadas y bajo sedación, en quirófano, pues son niños pequeños y no colaboran para una correcta exploración de la periferia de la retina. Es aconsejable obtener retinografías, que son de gran ayuda para el seguimiento, tanto para ver la evolución tras el tratamiento como para constatar que el resto tumoral está en regresión al no variar en exploraciones consecutivas, y así poder diferenciarlo de un tumor activo (Figura 10).
La ecografía ocular es una prueba de imagen inocua y fácil de realizar en los niños pequeños. Su indicación principal es para detectar calcificaciones intraoculares, un signo muy específico, aunque no patognomónico, del retinoblastoma, y también permite medir el tamaño tumoral. Hoy en día se considera que la ecografía ocular es la técnica más sensible para detectar calcio intraocular (Figura 11).
La resonancia magnética (RM) cerebral y orbitaria permite valorar la extensión del tumor a la órbita y el nervio óptico, y descartar un retinoblastoma trilateral. En la actualidad es la técnica de elección para estudiar la extensión extraocular del tumor, dado que es muy sensible para valorar la infiltración del nervio óptico y no emite radiaciones ionizantes. De todas formas, hemos de saber que la normalidad radiológica del nervio óptico no excluye la invasión histológica, sólo demostrable al enuclear el globo ocular (Figura 12).
Los modernos tomógrafos helicoidales, multicorte y multiplanares son muy rápidos, reducen la necesidad de anestesia en los niños y disminuyen la dosis de radiación. Delimitan muy bien las masas tumorales, con una sensibilidad mayor del 90% o 95% en la detección de calcificaciones. También permiten el estudio del nervio óptico y la diseminación del retinoblastoma a la órbita y al cráneo, pero puesto que emiten radiaciones ionizantes se han sustituido por la ecografía ocular y la RM orbitaria y cerebral, excepto en casos seleccionados.
La determinación enzimática en el humor acuoso, principalmente de lactato deshidrogenasa (LDH), sólo está indicada en caso de una gran duda diagnóstica. Aunque no es patognomónico, un cociente entre la LDH en el humor acuoso y el suero mayor de 1,5-2 sería sugestivo de retinoblastoma. Sólo se suele recurrir al estudio del humor acuoso cuando el retinoblastoma se presenta simulando una endoftalmitis o una uveítis, como ocurre en el retinoblastoma infiltrante difuso.
La biopsia vítrea está contraindicada por el riesgo de diseminar el tumor a través del lugar de la punción escleroconjuntival y empeorar el pronóstico vital del paciente. Si tuviera que plantearse, debería realizarse por vía anterior (córnea periférica, iris periférico, zónula, tumor) y sólo en aquellos casos en que sea imposible llegar al diagnóstico con métodos no invasivos.
El aspirado de la médula ósea, la punción lumbar y la gammagrafía ósea son pruebas para el estudio de la extensión extraocular. Las metástasis en el retinoblastoma son tardías, e improbables si el tumor es intraocular en el momento del diagnóstico. Por lo tanto, el estudio de extensión sólo se realiza cuando el retinoblastoma se diagnostica en un estadio muy avanzado, con extensión fuera del globo ocular, o si el estudio anatomopatológico tras la enucleación detecta tumor residual microscópico en el nervio óptico.
El retinoblastoma no es metabólicamente activo en su localización intraocular, por lo que la tomografía computarizada (TC)/tomografía de emisión de positrones (PET) no es útil para el control de las lesiones locales. En la órbita la resolución no es mayor que la de la RM, y además los músculos producen artefactos en la imagen. Tiene mucha utilidad en protocolos de extensión y detección precoz de metástasis a distancia.
Actualmente, una vez confirmado el diagnóstico, se obtiene sangre del paciente y de los padres y se realiza un estudio genético para estudio del gen RB1 y poder establecer si el retinoblastoma es germinal o somático, dado que esto permitirá ofrecer consejo genético y establecer la frecuencia de seguimiento del ojo contralateral en los casos unilaterales y la necesidad de seguimiento de los hermanos del paciente.
Una exploración oftalmológica exhaustiva puede aportar información importante que facilite el diagnóstico diferencial entre las causas de leucocoria. La realización de pruebas complementarias permite confirmar la presencia de calcio intralesional, altamente sugestiva de retinoblastoma, y permite excluir la mayoría de las causas de leucocoria. Así, la ecografía (en modos A y B) y la TC son de gran ayuda. La RM es muy útil para determinar la extensión extraocular del retinoblastoma, pero aporta menos información sobre las características del tumor intraocular.
Las principales patologías que pueden simular un retinoblastoma son:
Con menos frecuencia puede haber otras causas que den lugar a confusión diagnóstica con el retinoblastoma. Algunos tumores (meduloepitelioma, hemangioma coroideo, hamartoma combinado de retina y epitelio pigmentario…), malformaciones (colobomas coriorretinianos, pliegues congénitos retinianos…) y anomalías genéticas (incontinentia pigmenti, vitreorretinopatía exudativa familiar...), entre otras infrecuentes causas, pueden ocasionar un error diagnóstico que aboque en una enucleación evitable y en una angustia innecesaria para los familiares del niño. La exploración minuciosa por parte de un especialista en oftalmología pediátrica y la realización de las pertinentes pruebas complementarias puede minimizar el error diagnóstico y permitir un tratamiento precoz de estas afecciones (Figura 21 y Figura 22).
Debemos tener en cuenta:
La clasificación internacional para el estadiaje del retinoblastoma establece cinco estadios:
Clínica
El síntoma más frecuente es la leucocoria, seguida del estrabismo. Al inicio la leucocoria no es constante, y característicamente se observa en las fotografías con flash. El estrabismo, cuando ocurre, es constante con rapidez y refleja la pérdida visual del ojo afectado (Figura 1). Otros signos son rubeosis del iris, hipopion, hipema, heterocromía del iris, buftalmos, celulitis orbitaria y exoftalmos.
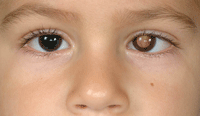
Figura 1. Leucocoria del ojo izquierdo en paciente con retinoblastoma unilateral.

La mayoría de los casos son detectados por el entorno familiar. Sólo un 5% a 7% se detectan en una exploración sistemática por un pediatra. La aparición de cualquiera de estos signos requiere la exploración del niño por un oftalmólogo, con examen de fondo de ojo.
Anatomía patológica
Macroscópicamente pueden observarse diferentes tipos de crecimiento tumoral: endofítico hacia la cavidad vítrea y exofítico a través de la retina hacia la coroides. También puede haber un patrón mixto endofítico y exofítico (Figura 2) y un patrón difuso.

Figura 2. Imagen macroscópica. Patrón de crecimiento mixto endofítico y exofítico.

En el examen histológico se aprecia que el retinoblastoma está constituido por células pequeñas, indiferenciadas, de núcleo basófilo e hipercromático, y escaso citoplasma (Figura 3). Se disponen difusamente y se agrupan formando rosetas de Flexner-Wintersteiner (Figura 4) constituidas por células cuboideas, de núcleo basal con proyecciones citoplasmáticas, dispuestas alrededor de una luz central, cuya identificación indica diferenciación. También puede verse alguna área con rosetas más diferenciadas, citológicamente benignas, con abundante citoplasma y núcleo pequeño, denominadas fleurettes (Figura 5).Tanto en los retinoblastomas diferenciados como en los indiferenciados pueden identificarse también rosetas de Homer-Wrigth; aunque éstas no son específicas del retinoblastoma, son semejantes a las rosetas de Flexner-Wintersteiner pero sin luz central.
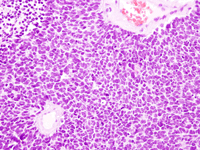
Figura 3. Células indiferenciadas de núcleo hipercromático y escaso citoplasma.

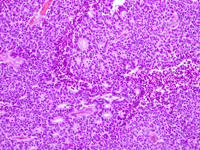
Figura 4. Rosetat de Flexner-Wintersteiner.


Figura 5. Numerosas fleurettes indicando diferenciación celular.

El retinoblastoma suele mostrar extensas zonas de necrosis, con presencia de calcificaciones; las zonas más preservadas son las que se encuentran alrededor de vasos sanguíneos (Figura 6). También suelen identificarse abundantes figuras de mitosis. Inmunohistoquímicamente las células tumorales expresan marcadores neurales, como enolasa neuronal específica, sinaptofisina y también proteína S100.

Figura 6. Necrosis tumoral. Células tumorales viables alrededor de estructuras vasculares.

La evaluación de la extensión del tumor es indispensable para definir los criterios histológicos de riesgo del retinoblastoma. Deben valorarse la invasión del nervio óptico, el margen de resección y la invasión poslaminar, intralaminar o prelaminar; también la invasión de coroides, de esclerótica y de la órbita, así como la extensión a la cámara anterior.
La evaluación de los criterios histológicos de riesgo ha variado considerablemente a lo largo del tiempo. En la actualidad (Tabla 1) se consideran criterios histológicos de bajo riesgo la invasión de la coroides y de la cámara anterior. Los criterios de riesgo intermedio son la invasión de la esclerótica intraescleral o la invasión del nervio óptico posterior a la lámina cribosa (Figura 7). Como criterios histológicos de alto riesgo se consideran la invasión de la esclerótica transescleral, la invasión del margen quirúrgico del nervio óptico (Figura 8) y la invasión extraocular.

Tabla 1. Criterios histológicos de riesgo del retinoblastoma.


Figura 7. Invasión del nervio óptico en la región poslaminar.

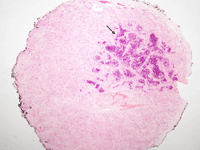
Figura 8. Invasión del margen quirúrgico del nervio óptico.

Por último, es muy importante realizar un informe de anatomía patológica estandarizado que incluya todos los hallazgos histológicos que permitan una correcta estadificación del tumor. Así, deben constar el diagnóstico, el patrón de crecimiento, la diferenciación, la invasión de estructuras y otros hallazgos histopatológicos que puedan encontrarse.
Diagnóstico
La oftalmoscopia indirecta es la técnica de elección para el diagnóstico del retinoblastoma. El resto de las pruebas complementarias ayudarán a descartar una afectación extraocular, a detectar calcificaciones intratumorales (muy útil en los casos de duda diagnóstica) y en el seguimiento postratamiento (Figura 9).

Figura 9. Algoritmo diagnóstico del retinoblastoma.

La exploración del fondo de ojo debe realizarse con las pupilas dilatadas y bajo sedación, en quirófano, pues son niños pequeños y no colaboran para una correcta exploración de la periferia de la retina. Es aconsejable obtener retinografías, que son de gran ayuda para el seguimiento, tanto para ver la evolución tras el tratamiento como para constatar que el resto tumoral está en regresión al no variar en exploraciones consecutivas, y así poder diferenciarlo de un tumor activo (Figura 10).

Figura 10. Retinoblastoma localizado nasal a la papila en el ojo izquierdo asociado a un desprendimiento exudativo de toda la retina.

La ecografía ocular es una prueba de imagen inocua y fácil de realizar en los niños pequeños. Su indicación principal es para detectar calcificaciones intraoculares, un signo muy específico, aunque no patognomónico, del retinoblastoma, y también permite medir el tamaño tumoral. Hoy en día se considera que la ecografía ocular es la técnica más sensible para detectar calcio intraocular (Figura 11).

Figura 11. Ecografía ocular. Se observa una gran masa intraocular que ocupa prácticamente toda la cavidad vítrea, con zonas hiperecogénicas intratumorales que corresponden a las calcificaciones.

La resonancia magnética (RM) cerebral y orbitaria permite valorar la extensión del tumor a la órbita y el nervio óptico, y descartar un retinoblastoma trilateral. En la actualidad es la técnica de elección para estudiar la extensión extraocular del tumor, dado que es muy sensible para valorar la infiltración del nervio óptico y no emite radiaciones ionizantes. De todas formas, hemos de saber que la normalidad radiológica del nervio óptico no excluye la invasión histológica, sólo demostrable al enuclear el globo ocular (Figura 12).

Figura 12. RM potenciada en T2. Se aprecia una masa hipointensa que ocupa prácticamente la totalidad de la cavidad vítrea del ojo derecho. No se observa afectación orbitaria ni cerebral, ni infiltración del nervio óptico.

Los modernos tomógrafos helicoidales, multicorte y multiplanares son muy rápidos, reducen la necesidad de anestesia en los niños y disminuyen la dosis de radiación. Delimitan muy bien las masas tumorales, con una sensibilidad mayor del 90% o 95% en la detección de calcificaciones. También permiten el estudio del nervio óptico y la diseminación del retinoblastoma a la órbita y al cráneo, pero puesto que emiten radiaciones ionizantes se han sustituido por la ecografía ocular y la RM orbitaria y cerebral, excepto en casos seleccionados.
La determinación enzimática en el humor acuoso, principalmente de lactato deshidrogenasa (LDH), sólo está indicada en caso de una gran duda diagnóstica. Aunque no es patognomónico, un cociente entre la LDH en el humor acuoso y el suero mayor de 1,5-2 sería sugestivo de retinoblastoma. Sólo se suele recurrir al estudio del humor acuoso cuando el retinoblastoma se presenta simulando una endoftalmitis o una uveítis, como ocurre en el retinoblastoma infiltrante difuso.
La biopsia vítrea está contraindicada por el riesgo de diseminar el tumor a través del lugar de la punción escleroconjuntival y empeorar el pronóstico vital del paciente. Si tuviera que plantearse, debería realizarse por vía anterior (córnea periférica, iris periférico, zónula, tumor) y sólo en aquellos casos en que sea imposible llegar al diagnóstico con métodos no invasivos.
El aspirado de la médula ósea, la punción lumbar y la gammagrafía ósea son pruebas para el estudio de la extensión extraocular. Las metástasis en el retinoblastoma son tardías, e improbables si el tumor es intraocular en el momento del diagnóstico. Por lo tanto, el estudio de extensión sólo se realiza cuando el retinoblastoma se diagnostica en un estadio muy avanzado, con extensión fuera del globo ocular, o si el estudio anatomopatológico tras la enucleación detecta tumor residual microscópico en el nervio óptico.
El retinoblastoma no es metabólicamente activo en su localización intraocular, por lo que la tomografía computarizada (TC)/tomografía de emisión de positrones (PET) no es útil para el control de las lesiones locales. En la órbita la resolución no es mayor que la de la RM, y además los músculos producen artefactos en la imagen. Tiene mucha utilidad en protocolos de extensión y detección precoz de metástasis a distancia.
Actualmente, una vez confirmado el diagnóstico, se obtiene sangre del paciente y de los padres y se realiza un estudio genético para estudio del gen RB1 y poder establecer si el retinoblastoma es germinal o somático, dado que esto permitirá ofrecer consejo genético y establecer la frecuencia de seguimiento del ojo contralateral en los casos unilaterales y la necesidad de seguimiento de los hermanos del paciente.
Diagnóstico diferencial
A pesar de que la clínica del retinoblastoma suele ser altamente sugestiva, no es infrecuente la confusión con otros cuadros. En un estudio de 2.775 pacientes remitidos al Wills Eye Hospital entre 1974 y 2011 con sospecha de retinoblastoma, 604 (22%) presentaban en realidad otro diagnóstico. Entre éstos, las causas más frecuentes fueron enfermedad de Coats (40%), persistencia de vascularización fetal (28%), hemovítreo (5%) y toxocariasis ocular (4%). En los niños menores de 1 año, la principal causa de error diagnóstico fue la persistencia de vascularización fetal (49%). En un estudio realizado en dos centros de Singapur sobre ojos enucleados por sospecha clínica de retinoblastoma, en tres de 28 casos se trató de otra etiología (dos casos de enfermedad de Coats y una toxocariasis ocular).Una exploración oftalmológica exhaustiva puede aportar información importante que facilite el diagnóstico diferencial entre las causas de leucocoria. La realización de pruebas complementarias permite confirmar la presencia de calcio intralesional, altamente sugestiva de retinoblastoma, y permite excluir la mayoría de las causas de leucocoria. Así, la ecografía (en modos A y B) y la TC son de gran ayuda. La RM es muy útil para determinar la extensión extraocular del retinoblastoma, pero aporta menos información sobre las características del tumor intraocular.
Las principales patologías que pueden simular un retinoblastoma son:
- Enfermedad de Coats: constituye la principal causa de error diagnóstico con el retinoblastoma. La presencia de vasos telangiectásicos en la periferia, la existencia de exudados lipídicos subretinianos y la ausencia de masa intraocular apoyan el diagnóstico de enfermedad de Coats. En ocasiones, un desprendimiento retiniano masivo puede dificultar el diagnóstico diferencial. Aunque algunos autores defienden la especificidad de los hallazgos en la RM en casos de duda diagnóstica, a veces estos hallazgos no han permitido la realización de un correcto diagnóstico diferencial. En los niños mayores, una angiografía fluoresceínica puede evidenciar la pérdida de colorante desde las telangiectasias retinianas periféricas (Figura 13 y Figura 14).

Figura 13. Enfermedad de Coats en un niño de 2 años de edad. Se observan la exudación lipídica masiva y las telangiectasias retinianas.


Figura 14. Enfermedad de Coats. Telangiectasias retinianas.

- Toxocariasis ocular: más frecuente en niños entre 2 y 6 años de edad, especialmente en contacto con perros o que hayan estado en contacto con tierra donde pueda haber heces de perro. La forma clínica de endoftalmitis crónica puede ocasionar dudas diagnósticas con el retinoblastoma infiltrativo difuso, y el granuloma periférico puede confundirse con un retinoblastoma único. La ausencia de calcio en las pruebas de neuroimagen y la presencia de celularidad en la cámara anterior y el vítreo anterior apoyan el diagnóstico de toxocariasis. La determinación de eosinofilia en sangre periférica, de utilidad en la toxocariasis sistémica, no ha demostrado ser útil en las formas oculares. La serología para Toxocara canis puede ser de utilidad, aunque algunos autores dudan de su efectividad y prefieren la determinación de anticuerpos antitoxocara en el humor acuoso (Figura 15 y Figura 16).

Figura 15. Toxocariasis ocular. Granuloma periférico con desprendimiento de retina total en una niña de 6 años de edad.


Figura 16. Toxocariasis ocular. Granuloma periférico.

- Persistencia de vascularización fetal: generalmente se presenta como una leucocoria diagnosticada al nacimiento o en las primeras semanas de vida. Sólo un 10% de los casos son bilaterales. La presencia de microftalmía, diámetro corneal reducido o elongación de los procesos ciliares es muy sugestiva de persistencia de vascularización fetal. La ecografía Doppler es de gran utilidad, pues permite descartar masas intraoculares y calcio, y evidenciar flujo sanguíneo a través de la arteria hialoidea (Figura 17).

Figura 17. Persistencia de vascularización fetal. Se observa una masa retrolental y tracción de los procesos ciliares.

- Hamartomas astrocíticos: son tumores de aspecto blanquecino, bien definidos, únicos o múltiples (en este caso generalmente asociados a esclerosis tuberosa). Suelen estar presentes al nacimiento y no crecer, aunque se han documentado casos de aparición de nuevos tumores y crecimiento de hamartomas previos. En ocasiones pueden calcificar y ocasionar confusión diagnóstica con el retinoblastoma. La presencia de otros tumores en la RM intracraneal o en otras localizaciones apoya el diagnóstico de hamartoma astrocítico. En casos de alta duda diagnóstica es necesario realizar fotografías de fondo de ojo seriadas para descartar el crecimiento rápido de las lesiones, que orientaría hacia el retinoblastoma (Figura 18 y Figura 19).

Figura 18. Hamartomas astrocíticos múltiples en un neonato afecto de rabdomiomas cardíacos en el contexto de una esclerosis tuberosa.


Figura 19. Hamartoma astrocítico con imagen cálcica en un niño con esclerosis tuberosa.

- Pars planitis: el retinoblastoma constituye el más temido síndrome mascarada en la edad pediátrica, pues puede simular una uveítis intermedia, en especial en los casos de retinoblastoma infiltrativo difuso. Una vitrectomía diagnóstica puede ser fatal debido al alto riesgo de diseminación local a través de las esclerotomías. La presencia de endotelitis, sinequias anteriores y la buena respuesta al tratamiento corticoideo apoyan el diagnóstico de pars planitis. En casos de alta duda diagnóstica puede ser de utilidad determinar la concentración de LDH en el humor acuoso y el cociente entre la LDH en éste y en suero. Valores altos son muy sugestivos, aunque no patognomónicos, de retinoblastoma (Figura 20).

Figura 20. Imagen RetCam de snowbank periférico en un niño afecto de parsplanitis, en quien se planteó el diagnóstico diferencial con un retinoblastoma infiltrativo difuso.

Con menos frecuencia puede haber otras causas que den lugar a confusión diagnóstica con el retinoblastoma. Algunos tumores (meduloepitelioma, hemangioma coroideo, hamartoma combinado de retina y epitelio pigmentario…), malformaciones (colobomas coriorretinianos, pliegues congénitos retinianos…) y anomalías genéticas (incontinentia pigmenti, vitreorretinopatía exudativa familiar...), entre otras infrecuentes causas, pueden ocasionar un error diagnóstico que aboque en una enucleación evitable y en una angustia innecesaria para los familiares del niño. La exploración minuciosa por parte de un especialista en oftalmología pediátrica y la realización de las pertinentes pruebas complementarias puede minimizar el error diagnóstico y permitir un tratamiento precoz de estas afecciones (Figura 21 y Figura 22).

Figura 21. Coloboma coriorretiniano inferior en un niño remitido para descartar un retinoblastoma.


Figura 22. Hamartoma mixto de retina-epitelio pigmentario.

Clasificación
Para la estadificación de los tumores intraoculares se usa la clasificación ABC del retinoblastoma (Tabla 2 y Figura 23, Figura 24, Figura 25, Figura 26, Figura 27, Figura 28 y Figura 29), en sustitución de la de Reese-Ellsworth, adaptada al tratamiento con radioterapia externa. Por otra parte, se ha propuesto una clasificación internacional que abarca desde los tumores intraoculares hasta la extensión extraocular.

Tabla 2. Clasificación del Retinoblastoma.


Figura 23. Estadio A de la clasificación ABC: dos tumores pequeños (flechas) respetando la mácula y el nervio óptico.


Figura 24. Grupo B de la clasificación ABC: tres tumores de diferentes tamaños cerca del nervio óptico y la mácula.


Figura 25. Grupo C de la clasificación ABC: tumor central en el polo posterior con siembras subretinianas localizadas.


Figura 26. Grupo C de la clasificación ABC: tumor con siembras vítreas localizadas.


Figura 27. Grupo D de la clasificación ABC: tumor con siembras vítreas masivas.


Figura 28. Grupo E de la clasificación ABC: tumor ocupando más del 50% del globo ocular.

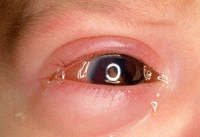
Figura 29. Grupo E de la clasificación ABC: tumor con invasión de la cámara anterior.

Debemos tener en cuenta:
- El carácter unilateral o bilateral de las lesiones.
- El número de tumores y su localización en la retina, en especial en relación a la mácula y el nervio óptico.
- El tamaño tumoral (diámetro y grosor).
- La presencia de fluido y de siembras subretinianas.
- La presencia y la localización de siembras vítreas.
La clasificación internacional para el estadiaje del retinoblastoma establece cinco estadios:
- Estadio 0: pacientes no enucleados.
- Estadio 1: pacientes enucleados con tumores completamente resecados.
- Estadio 2: pacientes enucleados con residuo microscópico.
- Estadio 3: enfermedad regional.
- Estadio 4: enfermedad metastásica (a) sin afectación del sistema nervioso central (SNC) y (b) con afectación del SNC.
Bibliografía recomendada
- Hosten N, Bornfeld N. Imaging of the globe and orbit; a guide to differential diagnosis. Stuttgart: Thieme; 1998. p. 182-191.
- Català-Mora J, Parareda-Salles A, et al. Síndrome mascarada por retinoblastoma difuso. Arch Soc Esp Oftalmol. 2009;84:477-80.
- Singh AD, Shields CL, Shields JA. Prognostic factors in retinoblastoma. J Pediatr Ophthalmol Strabismus. 2000;37:134-41.
- Finger PT, Harbour JW, Karcioglu ZA. Risk factors for metastasis in retinoblastoma. Surv Ophthalmol. 2002;47:1-16.
- Dunphy EB. The story of retinoblastoma. Am J Ophthalmol. 1964;58:539-52.
- Singh AD, Garway-Heath D, et al. Relationship of regression pattern to recurrence in retinoblastoma. Br J Ophthalmol. 1993;77:12-6.
- Shields CL, Palamar M, et al. Retinoblastoma regression patterns following chemoreduction and adjuvant therapy in 557 tumors. Arch Ophthalmol. 2009;127:282-90.
- Abramson DH, Gerardi CM, et al. Radiation regression patterns in treated retinoblastoma: 7 to 21 years later. J Pediatr Ophthalmol Strabismus.1991;28:108-12.
- Shields CL, Mashayekhi A, et al. Chemoreduction for retinoblastoma. Analysis of tumor control and risks for recurrence in 457 tumors. Am J Ophthalmol. 2004;138:329-37.
- Mashayekhi A, Shields CL, Eagle RC, Shields JA. Cavitary changes in retinoblastoma: relationship to chemoresistance. Ophthalmology. 2005;112:1145-50.
- Rojanaporn D, Kaliki S, et al. Intravenous chemoreduction or intra-arterial chemotherapy for cavitary retinoblastoma: long-term results. Arch Ophthalmol. 2012;130:585-90.
- Atchaneeyasakul L, Murphree AL. Retinoblastoma. En: Ryan SJ, ed. Retina. 3rd ed. St Louis: Mosby; 2001. p. 513.
- Ramon L, Font J, Croxatto O, Rao NA. Tumors of the Eye and Ocular Adnexa, Atlas of Tumor Pathology. 4rd series, Fascicle 5. Washington, DC: AFIP; 2006.p. 86-103.
- Abdel Razek AA, Elkhamary S, Al-Mesfer S, Alkatan HM. Correlation of apparent diffusion coefficient at 3T with prognostic parameters of retinoblastoma. AJNR. 2012;33(5):944-8.
- Razek A, Elkhamary S, Mousa A. Differentiation between benign and malignant orbital tumors at 3-T diffusion MR-imaging. Neuroradiology. 2011;53(7):517-22.
- Elkhamary S, Song K, et al. Can preoperative MR imaging predict optic nerve invasion of retinoblastoma? Eur J Radiol. 2012. http://dx.doi.org/10.1016/j.ejrad.2012.03.034.
- Ventura G, Cozzi G. Red reflex examination for retinoblastoma. Lancet. 2012; 380(9844):803.
- De Graaf P, Pouwels PJW, Rodjan F, et al. Single-shot turbo spin-echo diffusion-weighted imaging for retinoblastoma: initial experience. AJNR. 2012;33(1):110-8.
- Dimaras H, Kimani K, Dimba EA, et al. Retinoblastoma. Lancet. 2012;379(9824):1436-46.
- Abelairas-Gómez J, Peralta-Calvo J, Sánchez-Jacob E, Gayá-Moreno F. Introducción al retinoblastoma. Actualización en Cirugía Oftálmica Pediátrica. Capítulo 23. LXXVI Ponencia Oficial de la Sociedad Española de Oftalmología. 2000;23:265-76.
- De Graaf P, van der Valk P, Moll A, Imhof S, Schoutenvan A, Meeteren V, et al. Contrast-enhancement of the anterior eye segment in patients with retinoblastoma: correlation between clinical, MR imaging, and histopathologic findings. AJNR. 2010;31:237-45.
- Shields CL, Ghassemi F, Tuncer S, Thangappan A, Shelields JA. Clinical spectrum of diffuse infiltrating retinoblastoma in 34 consecutive eyes. Ophthalmology. 2008;115:2253-8.
- Saket RR, Mafee MF. Anterior-segment retinoblastoma mimicking pseudoinflammatory angle-closure glaucoma: review of the literature and the important role of imaging. AJNR. 2009;30:1607-9.
- Gorospe L, Royo A, Berrocal T, García-Raya P, Moreno P, Abelairas J. Imaging of orbital disorders in pediatric patients. Eur Radiol. 2003;13:2012-6.
- Chua J, Wisam J, Muen J, Reddy AJ. The masquerades of a childhood ciliary body medulloepithelioma: a case of chronic uveitis, cataract, and secondary glaucoma. Case Rep Ophthalmol Med. 2012;2012:493493.
- Mehta M, Sethi S, Pushker N, Kashyap S, Sen S, Bajaj MS, Ghose S. Retinoblastoma. Singapore Med J. 2012;53(2):128-36.
- Wilson MW, Rodriguez-Galindo C, Billups C, et al. Lack of correlation between the histologic and magnetic resonance imaging results of optic nerve involvement in eyes primarily enucleated for retinoblastoma. Ophthalmology. 2009;116:1558-63.
- Moll AC, Hoekstra OS, Imhof SM, et al. Fluorine-18 fluorodeoxyglucosepositron emission tomography (PET) to detect vital retinoblastoma in the eye: preliminary experience. Ophthalmic Genet. 2004;25:31-5.
- Dunkel IJ, Khakoo Y, Kernan NA, et al. Intensive multimodality therapy for patients with stage 4a metastatic retinoblastoma. Pediatr Blood Cancer. 2010;55:55-9.
- Brisse H, Guesmi M, Aerts I, Sastre-Garau X, Savignoni A, Lumbrosole Rouic L, et al. Relevance of CT and MRI in retinoblastoma for the diagnosis of postlaminar invasion with normal-size optic nerve: a retrospective study of 150 patients with histologic comparison. Pediatr Radiol. 2007;37:649-56.
- Silverman RH. High-resolution ultrasound imaging of the eye- a review. Clin Exp Ophthalmol. 2009;37:54-7.
- Song KD, Eo H, Kim JH, Yoo SY, Jeon TY. Can preoperative MR imaging predict optic nerve invasion of retinoblastoma? Eur J Radiol. 2012;81(12):4041-5.
- Finger PT, Meskin SW, Wisnicki HJ, et al. High frequency ultrasound of anterior segment retinoblastoma. Am J Ophthalmol. 2004;137:944-6.
- Shields CL, Mashayekhi A, Luo CK, et al. Optical coherence tomography in children: analysis of 44 eyes with intraocular tumors and simulating conditions. J Pediatr Ophthalmol Strabismus. 2004;41(6):338-44.
- Ruggeri M, Tsechpenakis G, Jiao S, et al. Retinal tumor imaging and volume quantification in mouse model using spectral-domain optical coherence tomography. Opt Express. 2009;17(5):4074-83.
- • Dhoot D, Weissman J, Landon R, Evans M, Stout T. Optic nerve enhancement in Coats disease with secondary glaucoma. J AAPOS. 2009;13:301-2.
- Galluzzi P, Hadjistilianou T, Cerase A, De Francesco S, Toti P, Venturi C. Is CT still useful in the study protocol of retinoblastoma? AJNR. 2009;30(9):1760-5.
- Lemke A, Kazi I, Mergner U, Foerster P, Heimann H, Bechrakis N, et al. Retinoblastoma—MR appearance using a surface coil in comparison with histopathological results. Eur Radiol. 2007;17:49-60.
- De Graaf P, Knol D, Moll A, Imhof S, Schouten-van Meeteren A, Castelijns J. Eye size in retinoblastoma: MR imaging measurements in normal and affected eyes. Radiology. 2007;244:273-80.
- Lope L, Hutcheson K, Khademian Z. Magnetic resonance imaging in the analysis of pediatric orbital tumors: utility of diffusion-weighted imaging. JAAPOS. 2010;14(3):257-62.
- Dai S, Dimaras H, Héon E, et al. Trilateral retinoblastoma with pituitary-hypothalamic dysfunction. Ophthalmic Genetics. 2008;29(3):120-5.
- Abramson D, Beaverson C, Sangani P, et al. Screening for retinoblastoma: presenting signs as prognosticators of patient andocular survival. Pediatrics. 2003;112:1248-55.
- Chantada G, Qaddoumi I, Canturk S, et al. Strategies to manage retinoblastoma in developing countries. Pediatr Blood Cancer. 2011;56:341-8.
- Lucignani G, De Palma D. PET/CT in paediatric oncology: clinical usefulness and dosimetric concerns. Eur J Nucl Med Mol Imaging. 2011;38:179-84.
- Radhakrishnan V, Kumar R, Arun Malhotra A, Bakhshil S. Role of PET/CT in staging and evaluation of treatment response after 3 cycles of chemotherapy in locally advanced retinoblastoma: a prospective study. J Nucl Med. 2012;53:191-8.
- Dunkel IJ, Jubran RF, Guruangan S, et al. Trilateral retinoblastoma: potentially curable with intensive chemotherapy. Pediatr Blood Cancer. 2010;54(3):384-7.
- Gatta G, Capoaccia R, Stiller C, et al. Childhood cancer survival trends in Europe: a EUROCARE Working Group Study. J Clin Oncol. 2005;23:3742-51.
- Canturk S, Qaddoumi I, Khetan V, et al. Survival of retinoblastoma in less-developed countries impact of socioeconomic and health-related indicators. Br J Ophthalmol. 2010;94:1432-6.
- Stevenson KE, Hungerford J, Gardner A. Local extraocular extension following intraocular surgery. Br J Ophthalmol. 1989;73:739-42.
- Shields CL, Honavar S, Shields JA, Demirci H. Vitrectomy in eyes with unsuspected retinoblastoma. Ophthalmology. 2000;107:2250-5.
- Palma J, Sasso DF, Dufort G, Koop K, Sampor C, Diez B, et al. Bone successful treatment of metastatic retinoblastoma with high-dose chemotherapy and autologous stem cell rescue in South America. Bone Marrow Transplant. 2012;47(4):522-7.
- Abramson DH, Beaverson C, Sangani P, et al. Screening for retinoblastoma: presenting signs as prognosticators of patient and ocular survival. Pediatrics. 2003;112:1248-55.
- Shields JA, Shields CL. Management of retinoblastoma. En: Intraocular tumors: An atlas and textbook. 2nd ed. Philadelphia: Lippincott Williams and Wilkins; 2008.p. 334-51.