Volumen 20 - Número 2 - Abril-Junio 2012
Ependimoma en síndrome de Noonan
P. Rocha1, JA. Abreu2, CN. Hernández3, R. López4, H. Roldán5
1Licenciado en Medicina. Servicio de Oftalmología
2Doctor en Medicina. Servicio de Oftalmología
3Licenciado en Medicina. Servicio de Anatomía Patológica
4Licenciado en Medicina. Servicio de Pediatría
5Doctor en Medicina. Servicio de Neurocirugía
2Doctor en Medicina. Servicio de Oftalmología
3Licenciado en Medicina. Servicio de Anatomía Patológica
4Licenciado en Medicina. Servicio de Pediatría
5Doctor en Medicina. Servicio de Neurocirugía
CORRESPONDENCIA
Pedro Rocha Cabrera
Calle Santo Domingo nº26 2º Dcha.
38003 Santa Cruz de Tenerife, España
E-mail: procha975@yahoo.es
Calle Santo Domingo nº26 2º Dcha.
38003 Santa Cruz de Tenerife, España
E-mail: procha975@yahoo.es
RESUMEN
Caso clínico: Varón de 13 años diagnosticado de Síndrome de Noonan (SN) remitido a nuestro servicio por presentar cefalea. En la exploración ocular se observa papiledema bilateral. La RMN muestra la existencia de una neoformación intracraneal en fosa posterior. El estudio histopatológico confirmó el diagnóstico de ependimoma.
Discusión: Entre la patología tumoral descrita asociada al SN están los síndromes mieloproliferativos, neoplasia de células gigantes maxilar superior y mandibular, y el rabdomiosarcoma. En la literatura no hemos encontrado descrita la asociación de ambas patologías, por lo que en nuestro caso pudiera tratarse de una variante del SN o de una presentación casual.
Discusión: Entre la patología tumoral descrita asociada al SN están los síndromes mieloproliferativos, neoplasia de células gigantes maxilar superior y mandibular, y el rabdomiosarcoma. En la literatura no hemos encontrado descrita la asociación de ambas patologías, por lo que en nuestro caso pudiera tratarse de una variante del SN o de una presentación casual.
RESUM
Cas clínic: Home de 13 anys diagnosticat de Síndrome de Noonan (SN) remès al nostre Server per presentar cefalea. A l'exploració ocular s'observa papiledema bilateral. La RMN mostra l'existència d'una neoformació intracraneal en fossa posterior. L'estudi histopatològic va confirmar el diagnòstic d'ependimoma.
Discussió: Entre la patologia tumoral descrita associada al SN estan les síndromes
mieloproliferatives, neoplàsia de cèl•lules gegants maxil•lar superior i mandibular,
i el rabdomiosarcoma. A la literatura no hem trobat descrita l'associació d'ambdues patologies, pel que en el nostre cas podria tractar-se d'una variant del SN o d'una presentació casual.
Discussió: Entre la patologia tumoral descrita associada al SN estan les síndromes
mieloproliferatives, neoplàsia de cèl•lules gegants maxil•lar superior i mandibular,
i el rabdomiosarcoma. A la literatura no hem trobat descrita l'associació d'ambdues patologies, pel que en el nostre cas podria tractar-se d'una variant del SN o d'una presentació casual.
ABSTRACT
Case report: 13 year old male diagnosed with Noonan syndrome (NS) was referred to our department with headache. The eye examination shows bilateral papilledema. MRI showed the existence of an intracranial neoplasm in the posterior fossa. The histopathological examination confirmed the diagnosis of ependymoma.
Discussion: Among the pathology associated with NS tumor are described myeloproliferative syndromes, giant cell tumor of the maxilla and mandible, and rhabdomyosarcoma. In the literature we have not found described the association of both disorders, so in our case it could be a variant of the NS or a casual presentation.
Discussion: Among the pathology associated with NS tumor are described myeloproliferative syndromes, giant cell tumor of the maxilla and mandible, and rhabdomyosarcoma. In the literature we have not found described the association of both disorders, so in our case it could be a variant of the NS or a casual presentation.
Introducción
El Síndrome de Noonan (SN) es una enfermedad autosómica dominante, en ocasiones esporádica, con una prevalencia de uno de cada 2.500 recién nacidos vivos. Se caracteriza por talla corta, dismorfias craneofaciales (hipertelorismo, proptosis, ptosis palpebral, hipoplasia mandibular, implantación baja de las orejas y del pelo de la nuca, pterigium colli), anomalías torácicas y cardíacas (estenosis pulmonar)1, malformaciones en los dedos de las manos y de los pies, retraso mental en el 25% de los casos, criptorquidia, hipoacusia neurosensorial y alteraciones en la coagulación. La patología tumoral relacionada con el SN son los síndromes mieloproliferativos2, neoplasias de células gigantes en maxilar superior y mandíbula3, y rabdomiosarcomas en diferentes localizaciones4.El ependimoma es un tumor de crecimiento lento de mayor incidencia en niños y adultos jóvenes, tercero en frecuencia de los localizados en la fosa posterior intracraneal5, siendo el 4% de todos lo tumores cerebrales de la primera década6. La media de edad en el diagnóstico tumoral se encuentra entre los 4-6 años de edad7,8. El 90% de estos tumores se localizan en el cerebro, y el otro 10% se ubican en la espina dorsal especialmente en adultos. La sintomatología depende de la localización primaria del tumor, así podemos encontrarlos en la fosa posterior, en el área supratentorial y en la espinal dorsal9. Los tumores de la fosa posterior pueden producir obstrucción de la circulación del líquido cefalorraquídeo (LCR) y originar hipertensión intracraneal (HIC), estando los del cuarto ventrículo asociados a tortícolis y ataxia. Los tumores de la espina dorsal cursan con dolor de espalda, ciatialgia, debilidad y asimetría en miembros inferiores y escoliosis.
Caso clínico
Varón de 13 años diagnosticado de SN, con mutación en el gen PTPN11, en tratamiento con hormona de crecimiento (GH) desde hace 3 meses, remitido a nuestro servicio por cefalea pancraneal, evidenciándose en la exploración ocular papiledema; el estudio del campo visual (CV) muestra afectación “sin patrón” de predominio en hemicampos izquierdos (Figuras 1a y 1b). El servicio de Pediatría realiza un diagnóstico inicial de HIC benigna posiblemente relacionada con la administración de GH, por lo que es suspendida. A la semana acude al servicio de urgencias por un episodio de cefalea acompañada de vómitos en escopetazo y ataxia. Se “agiliza” la realización estudios de neuroimagen (TAC y RMN) solicitados previamente, observándose en los mismos la presencia de hidrocefalia y de una tumoración en fosa posterior (Figura 2). El servicio de Neurocirugía realiza una derivación ventrículo-peritoneal y extirpación del tumor. Como secuela postquirúrgica el paciente presenta paresia del III par bilateral y paresia del VI par izquierdo. El estudio histopatológico del material extraído confirma el diagnóstico de ependimoma grado II OMS (Figura 3).
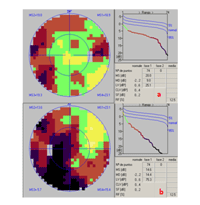
Figura 1. Estudio inicial de CV de ambos ojos (programa 32 estrategia TOP). a) O.D. Se observa afectación sin patrón de predominio en hemicampo izquierdo. CV inicial OD. b) Misma efactación en OI
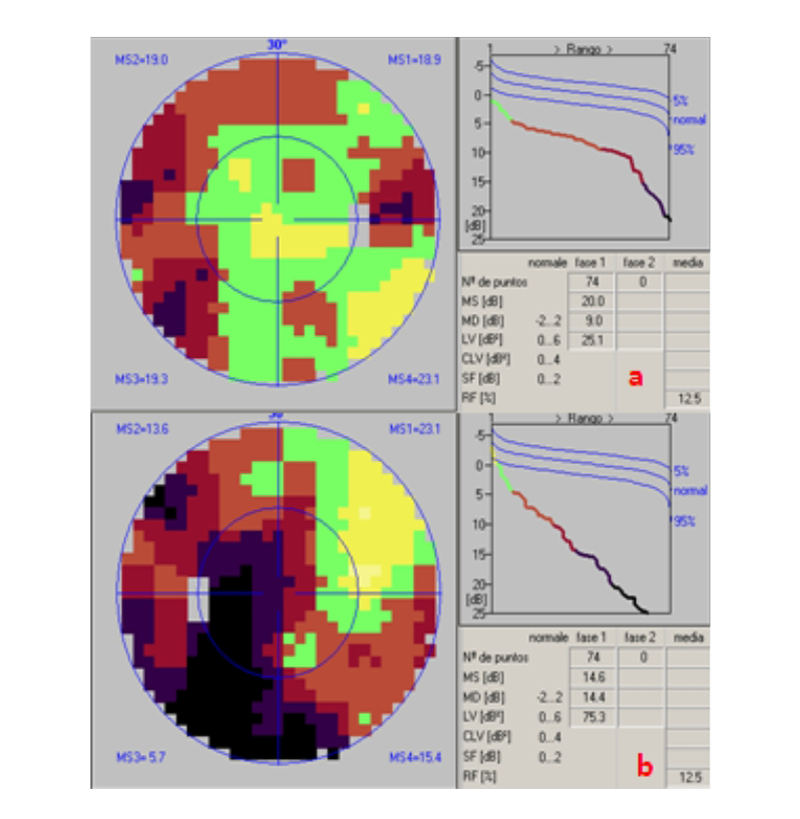
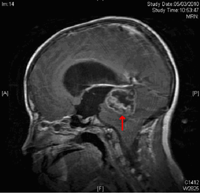
Figura 2. RMN T1 sagital inicial tras administración de contraste con Gadolinio: se observa tumoración que capta contraste en la periferia pero no en el centro debido a la necrosis tumoral, se observa la presencia de gran dilatación ventricular, como se observa a nivel de la silla turca
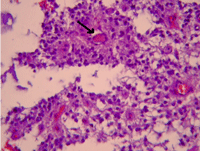
Figura 3. Estudio histopatológico: se observa la existencia de "seudorosetas" perivasculares (tinción Hematoxilina-Eosina)
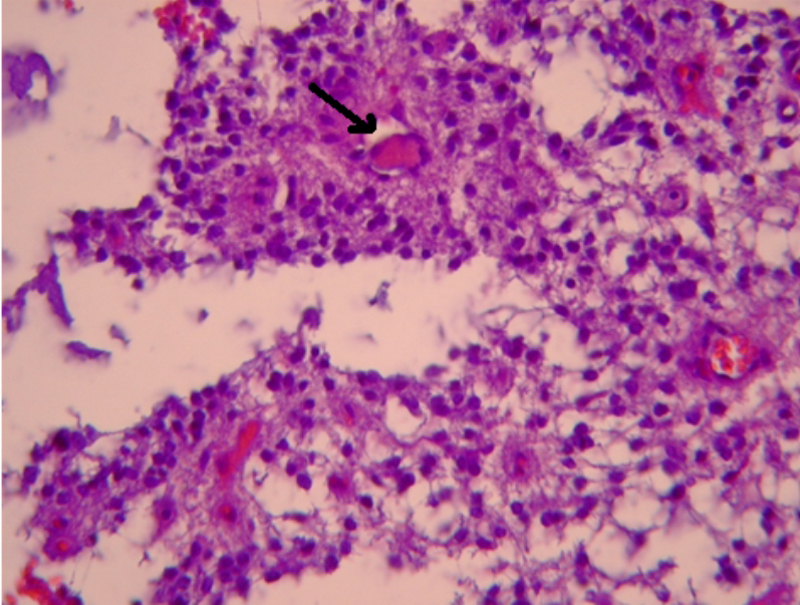
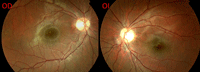
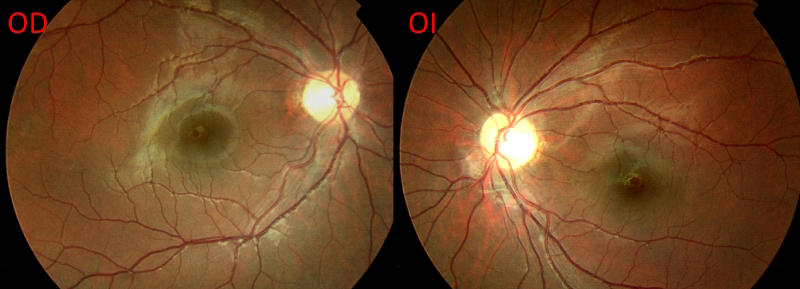
Figura 4. Retinografía actual: se observa palidez papilar bilateral
En la RMN de control se observan dos áreas de captación patológica de Gadolinio, una en la vertiente superior y lateral izquierda de 1 cm y otra en la vertiente anterior y lateral derecha del cuarto ventrículo, por lo que el paciente es remitido al hospital de referencia (Hospital Vall d´Hebron, Barcelona), para realizarle tratamiento radioterápico focal con fraccionamiento diario de 1.8 Gy y dosis total de 56 Gy. En la actualidad se aprecia una afectación bilateral incompleta del III par con leve ptosis, VI par izquierdo, sin diplopía en posición primaria de la mirada, palidez papilar (Figura 4), test de visión binocular negativos, mejoría franca del CV, y estudios de neuroimagen sin evidencia de hidrocefalia ni del tumor.
Discusión
La patología tumoral descrita hasta el momento asociada al SN son los síndromes mieloproliferativos, neoplasia de células gigantes maxilar superior y mandibular, y el rabdomiosarcoma; en nuestro paciente se observa un ependimoma.En el diagnóstico diferencial de la cefalea en la infancia debemos tener en cuenta procesos infecciosos generales, fiebre, crisis migrañosa, sinusitis aguda, meningitis, encefalitis, hematoma subdural, tumor intracraneal como sucedió en nuestro caso, absceso cerebral, hemorragia subaracnoidea, neuritis óptica, epilepsia, hidrocefalia aguda y HIC benigna, primer diagnóstico realizado por el servicio de pediatría, sospechado por el uso de GH10. Se cree que la GH tiene una acción de IGF-I-mediada por el plexo coroideo, principal lugar de producción de LCR, aumentando la producción del mismo.
El tratamiento de elección del ependimoma es la cirugía, mejorando el pronóstico cuando la exéresis es completa11,12, no siempre posible dado que en el 30-40% de los casos el tumor invade estructuras importantes12,13, como ocurrió en nuestro caso. En los pacientes con una completa resección tumoral la supervivencia a los cinco años es del 67-80%, siendo del 22-47% cuando no lo es14.
Como tratamiento coadyuvante a la cirugía debe realizarse tratamiento con radioterapia aunque la misma sea completa, especialmente en niños mayores de 3 años15 para evitar posibles recidivas16,17. El uso de quimioterapia es controvertido y existen indicios de resistencia del ependimoma a la misma18, no mejorando la supervivencia tras su aplicación19.
La OMS clasifica el ependimoma en cuatro tipos histológicos: mixopapilar, subependimomas, ependimomas y ependimomas anaplásicos11, y en tres grados en función de la agresividad tumoral (del I al III). Nuestro paciente presenta un ependimoma grado II, que se caracteriza por la presencia de “seudorosetas” perivasculares, siendo poco frecuente la presencia de rosetas verdaderas; se reconocen núcleos redondeados uniformes sobre un fondo fibrilar con nucleolo visible. Son lesiones bien delimitadas histológicamente19.
La diseminación cerebroespinal se presenta aproximadamente en el 13% de los pacientes8. Los ependimomas recidivan localmente cuando la exéresis no es completa, haciéndolo raramente a distancia.
Entre los criterios de exclusión recomendados para el uso de GH se encuentra la existencia de un proceso tumoral activo, debiéndose determinar T4 libre y anticuerpos antitiroideos, IGF-1 e IGFBP3, hemoglobina glicosilada, y marcadores de enfermedad celíaca20. En nuestro paciente la RMN fue realizada tras presentar clínica de HIC.
La asociación de un ependimoma al SN no la hemos encontrado descrita en la literatura revisada, por lo que nuestro caso pudiera tratarse de una variante del mismo o de una presentación casual.
Bibliografía
- Aypar E, Atalay S, Tutar E, Demir F. Unusual cardiac phenotype in a newborn with Noonan syndrome. Congenit Heart Disease. 2010;5(2):178-81.
- Hasle H. Malignant diseases in Noonan syndrome and related disorders. Hormone Research in Paediatrics. 2009;72 (Suppl. 2):8-14.
- Bufalino A, Carrera M, Carlos R, Coletta RD. Giant Cell Lesions in Noonan Syndrome: case report and review of the Literature. Head Neck Pathology. 2010; 4(2):174-7.
- Moschovi M, Touliatou V, Papadopoulou A, Mayakou MA, Nikolaidou-Karpathiou P, Kitsiou-Tzeli S. Rhabdomyosarcoma in a patient with Noonan syndrome phenotype and review of the literature. Journal Pediatric Hematology/ Oncology. 2007; 29(5):341-4.
- Osborn AG, Blazer SI, Salzman KL. Serie radiología clínica. Los 100 diagnósticos principales en cerebro. Madrid: Elsevier, 2007; p. 150.
- Bradley WG, Daroff RB, Fenichel GM, Jankovic J. Neurology in clinical practice. Fourth Edition. Philadelphia: Elsevier, 2004;II: 1352.
- Foreman NK, Love S, R T. Intracranial ependymomas: Analysis of prognostic factors in a population-based series. Pediatric Neurosurgery. 1996;24:119-25.
- Horn B, Heideman R, Geyer R, et al. A multi-institutional retrospective study of intracranial ependymoma in children: Identification of risk factors. Journal Pediatric Hematology Oncology. 1999;21:203-11.
- Wiestler O, Schiffer D, Coons S, Prayson R, Rosenblum M. Ependymal Tumors. En: Kleihues P, Cavenee W, eds. Tumors of the Central Nervous System. Lyon (France): IARC Press, 2000:72-82.
- Domínguez M, Santiago R, Campos J, Férnandez de Péres MJ. La cefalea en la infancia. Anales Esp. Pediatría. 2002;57(5):432-43.
- Healey E, Barnes P, Kupsky W, et al. The prognostic significance of post-operative residual tumor in ependymoma. Neurosurgery. 1991;28:666-72.
- Nazar G, Hoffman H, Becker L, et al. Infratentorial ependymomas in childhood; prognostic factors and treatment. J Neurosurg. 1990;72:408-17.
- Duffner P. Postoperative chemotherapy and delayed radiation in children less than three years of age with malignant brain tumors. New England Journal of Medicine. 1993;328:1725-31.
- Merchant T. Current Management of Childhood Ependymoma. Oncology. 2002; 16:629-44.
- Paulino A. Radiotherapeutic Management of Intracranial Ependymoma. Pediatric Hematology & Oncology. 2002;19:295-308.
- Mork SJ, AC L. Ependymoma: A follow-up study of 101 cases. Cancer. 1977; 40:907-15.
- Shuman RM, Alvord EC, RW L. The biology of childhood ependymomas. Arch Neurol. 1975;32:731-9.
- Chou P, Barquin N, Gonzalez-Crussi F, Tomita T, Reyes-Mugica M. Ependymomas in children express the multidrug-resistance gene: immunohistochemical and molecular biologic study. Fetal and Pediatric Pathology. 1996;16:551-61.
- Rubien P. Oncología clínica. Octava edición. Elsevier: Madrid, 2003:797-813.
- Ministerio de Sanidad y Consumo. Protocolos. Disponible en: http://www.msc.es/profesionales/farmacia/pdf/criteriosHCninos020908.pdf (fecha consulta 24 abril 2011).